Gebrauchsanweisung für Medizinprodukte
27.3.2020 Gesetzgebung
Die Erstellung von Gebrauchsanweisungen für Medizinprodukte geht mit recht viel Verantwortung einher. In der Tat müssen Sie Ihre technischen Fähigkeiten mit Projektmanagement-, Verwaltungs- und Rechtskenntnissen kombinieren. Glücklicherweise wurde in den einschlägigen Gesetzen festgelegt, was in die Gebrauchsanweisungen für Medizinprodukte aufzunehmen ist. Wenn Sie Gebrauchsanweisungen für Medizinprodukte erstellen möchten, müssen Sie sowohl den Inhalt als auch den zu befolgenden Prozess berücksichtigen. In diesem Leitfaden werde ich Ihnen alles Wissenswerte aufzeigen, was Sie zur Erstellung von Gebrauchsanweisungen für Medizinprodukte benötigen. Ich werde Ihnen zeigen, wie Sie solche Gebrauchsanweisungen für den europäischen Markt sowie für den US-Markt erstellen.
Wenn es eine Branche gibt, in der es wichtig ist, sicherzustellen, dass die Anwender Produkte sicher, effektiv und effizient verwenden können, dann ist dies die Medizinproduktebranche.
Und wir dürfen auch nicht vergessen, wie wichtig es ist, dass Sie Ihre Haftung durch angemessene Gebrauchsanweisungen verringern.
Los geht’s!
HABEN SIE KEINE ZEIT, DEN GANZEN ARTIKEL
ZU LESEN? LADEN SIE DIE ANFORDERUNGEN AN
GEBRAUCHSANWEISUNGEN FÜR MEDIZINPRODUKTE HERUNTER

Über die Verordnung (EU) 2017/745
Die Verordnung (EU) 2017/745 ersetzt die Richtlinie 93/42/EWG über Medizinprodukte 93/42/EWG. Sie ersetzt auch die Richtlinie 90/385/EWG über aktive implantierbare medizinische Geräte.
Die neue Medizinprodukteverordnung tritt am 26. Mai 2020 in Kraft.
Zunächst einmal: Warum ist die Richtlinie zu einer Verordnung geworden und was ist der Unterschied zwischen den beiden?
Eine Verordnung ist in allen Mitgliedsstaaten des Europäischen Wirtschaftsraums (EWR) rechtsverbindlich. Eine Richtlinie legt bestimmte Ergebnisse fest, die erzielt werden müssen. Jeder einzelne Mitgliedstaat entscheidet jedoch selbst, wie er die Richtlinien in nationales Recht umsetzt.
Die Verordnung harmonisiert die Vorschriften für das Inverkehrbringen und die Inbetriebnahme von Medizinprodukten und deren Zubehör auf dem EU-Markt.
Ziel der Verordnung ist es, den freien Verkehr sicherer Medizinprodukte innerhalb der EU zu gewährleisten.
Aber was genau ist ein Medizinprodukt?
Ein Medizinprodukt ist ein Instrument, ein Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder ein anderes Objekt, das vom Hersteller dafür vorgesehen ist, allein oder in Kombination für einen oder mehrere der folgenden spezifischen medizinischen Zwecke am Menschen verwendet zu werden:
- Diagnose, Prävention, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung oder Kompensation einer Verletzung oder Behinderung,
- Untersuchung, Ersetzung oder Änderung der Anatomie oder eines physiologischen oder pathologischen Prozesses oder Zustands,
- Bereitstellung von Informationen durch In-vitro-Untersuchung von Proben aus dem menschlichen Körper, einschließlich Organ-, Blut- und Gewebespenden.
Die Verordnung schreibt vor, dass alle diese Produkte:
- für den vorgesehenen Zweck geeignet und
- sicher und effektiv sein müssen sowie
- den klinischen Zustand oder die Sicherheit der Patienten oder die Sicherheit und Gesundheit der Anwender nicht beeinträchtigen dürfen.
Die Verordnung gilt auch für Zubehör für Medizinprodukte und für bestimmte Produktgruppen ohne medizinischen Zweck, wie z. B. Kontaktlinsen, Produkte, die dazu bestimmt sind, durch chirurgische Eingriffe ganz oder teilweise in den menschlichen Körper eingeführt zu werden, Produkte, die dazu bestimmt sind, die Anatomie zu verändern oder zur Fixierung von Körperteilen dienen, Geräte, die elektromagnetische Strahlung hoher Intensität aussenden, und Geräte, die zur Hirnstimulation bestimmt sind.
Die Verordnung gilt nicht für In-vitro-Diagnostika, Arzneimittel, Kosmetikprodukte, menschliches Blut, Transplantate und Lebensmittel.
MÖCHTEN SIE EINE VORLAGE FÜR EINE GEBRAUCHSANWEISUNG NUTZEN, DIE BEREITS ALLE RECHTLICHEN ASPEKTE ENTHÄLT?
Nehmen Sie den schnellsten Weg zu einer konformen Anleitung. Wir haben Vorlagen für Gebrauchsanweisungen für Medizinprodukte entwickelt, die bereits alle rechtlichen Inhalte beinhalten.
Klassifizierung: Liste der Medizinprodukteklassen
Medizinprodukte wurden durch die Verordnung in mehrere Klassen unterteilt:
- Klasse I
- Klasse IIa
- Klasse IIb
- Klasse III
Eine Benannte Stelle, auch Notifizierte Stelle genannt, muss an der Bewertung von Medizinprodukten der Klassen IIa, IIb und III gemäß der Medizinprodukteverordnung beteiligt sein. Die Gebrauchsanweisung ist Teil des Konformitätsbewertungsverfahrens. Sie wird mindestens auf Vollständigkeit und gelegentlich auch auf Richtigkeit geprüft.
Die Gebrauchsanweisung muss Bestandteil eines Medizinproduktes sein. In Ausnahmefällen sind für Produkte der Klassen I und IIa keine Gebrauchsanweisungen erforderlich, wenn solche Produkte ohne solche Anweisungen sicher verwendet werden können.
Aus diesem Grund bestimmt die Klassifizierung Ihres Produkts auch, ob und wie Sie Ihre Gebrauchsanweisung erstellen sollten.
Welche Informationen sollten in der Gebrauchsanweisung für Medizinprodukte enthalten sein?
Höchstwahrscheinlich wissen Sie inzwischen, dass Sie ein Medizinprodukt haben, für das eine Gebrauchsanweisung erforderlich ist. Die nächste Frage wäre: Welche Informationen muss Ihre Gebrauchsanweisung enthalten?
Dieses Video zeigt Ihnen, wie Sie die rechtliche Anforderungen für Ihre Medzinprodukte bestimmen:
Die Verordnung enthält die folgende (leicht umformulierte und umstrukturierte) Liste:
- Name oder Handelsname des Medizinprodukts;
- Name oder Handelsname (oder eingetragene Marke) und Anschrift des Herstellers;
- Ausstellungsdatum der Gebrauchsanweisung oder Überarbeitungsdaten und Informationen darüber, wo die letzte Überarbeitung der Gebrauchsanweisung zu finden ist;
- Gegebenenfalls den Hinweis, dass das Medizinprodukt ein Arzneimittel (einschließlich eines Derivats aus menschlichem Blut oder Plasma), Gewebe oder Zellen menschlichen Ursprungs (oder ihre Derivate) oder Gewebe oder Zellen tierischen Ursprungs (oder ihre Derivate) enthält;
- Gegebenenfalls Angaben zu Stoffen, die kennzeichnungspflichtig sind (gemäß Abschnitt 10.4.5 der Verordnung);
- Wenn das Medizinprodukt steril geliefert, einen Hinweis auf den sterilen Zustand und das Sterilisationsverfahren;
- Gegebenenfalls Anweisungen hinsichtlich des Vorgehens für den Fall, dass die Sterilverpackung vor der Verwendung des Produkts beschädigt oder versehentlich geöffnet wurde;
- Anleitungen zur Sterilisation (gilt nur für Medizinprodukte, die nicht steril geliefert werden und vor Gebrauch sterilisiert werden müssen);
- Gegebenenfalls einen Hinweis darauf, dass das Medizinprodukt zum einmaligen Gebrauch bestimmt ist, wenn dies der Fall ist;
- Bei Produkten, die aus Stoffen bestehen, die dazu bestimmt sind, durch eine Körperöffnung oder durch Anwendung auf der Haut in den menschlichen Körper eingeführt zu werden, und die vom menschlichen Körper aufgenommen oder lokal im Körper verteilt werden, die qualitative Gesamtzusammensetzung des Produkts und quantitative Informationen zu dem(n) Hauptbestandteil(en), der (die) für das Erreichen der angestrebten Hauptwirkung verantwortlich ist (sind);
- Die Zweckbestimmung des Produkts einschließlich Kontraindikationen, eine genaue Angabe der Indikationen, Patientenzielgruppe(n) und vorgesehenen Anwender;
- Informationen über besondere Einrichtungen sowie besondere Schulungen oder spezifische Qualifikationen des Produktanwenders und/oder Dritter;
- Informationen zu Lagerung und/oder Handhabung;
- Informationen, die erforderlich sind, um zu überprüfen, ob das Medizinprodukt ordnungsgemäß installiert wurde und zur bestimmungsgemäßen Verwendung bereit ist. Diese Informationen müssen gegebenenfalls Anleitungen zur Instandhaltung, Reinigung/Desinfektion, zum Austausch von Verbrauchsmaterialien, zur Kalibrierung und zur Beseitigung von Risiken enthalten;
- Detaillierte Informationen zu allen vorbereitenden Behandlungen oder der Handhabung des Produkts, bevor es einsatzbereit ist oder während es verwendet wird (z. B. Sterilisation, Kalibrierung, Endmontage). Außerdem müssen die erforderlichen Desinfektionsniveaus und die verfügbaren Methoden zum Erreichen dieser Niveaus angegeben werden.
- Gegebenenfalls Angaben zu dem zu erwartenden klinischen Nutzen;
- Gegebenenfalls Links zum Kurzbericht über Sicherheit und klinische Leistung (siehe Artikel 32 der Verordnung);
- Die Leistungsmerkmale des Medizinprodukts;
- Gegebenenfalls Angaben, anhand deren ein Angehöriger der Gesundheitsberufe überprüfen kann, ob das Produkt geeignet ist.
- Gegebenenfalls Angaben anhand deren ein Angehöriger der Gesundheitsberufe die entsprechende Software und die entsprechenden Zubehörteile auswählen kann;
- Alle Restrisiken, Kontraindikationen und unerwünschte Nebenwirkungen. Dies muss Informationen enthalten, die dem Patienten mitzuteilen sind.
- Alle relevanten Warnungen, Vorsichtsmaßnahmen, Kontraindikationen, zu ergreifende Maßnahmen und Verwendungsbeschränkungen in Bezug auf das Produkt.
- Alle relevanten Warnungen und Vorsichtsmaßnahmen, wenn Substanzen in den menschlichen Körper eingeführt werden sollen;
- Angaben, die der Anwender benötigt, um das Medizinprodukt angemessen zu verwenden wie z. B. der für das Produkt geltende Genauigkeitsgrad, wenn es eine Messfunktion hat;
- Bei wiederverwendbaren Produkten Angaben zu den Verfahren zur Reinigung, Desinfektion, Verpackung und gegebenenfalls über das validierte Verfahren zur erneuten Sterilisation entsprechend dem Land, in dem das Produkt in Verkehr gebracht worden ist. Es muss erwähnt werden, dass das Produkt nicht mehr verwendet werden darf, wenn es zum Beispiel Anzeichen für eine Materialabnutzung gibt oder die maximal zulässige Anzahl von Wiederverwendungen erreicht wurde;
- Gegebenenfalls einen Hinweis, dass ein Medizinprodukt wiederverwendet werden kann, wenn es unter der Verantwortung des Herstellers aufbereitet worden ist;
- Bei Produkten, die nur zum einmaligen Gebrauch bestimmt sind, müssen Informationen über die Risiken enthalten sein, die auftreten können, wenn das Produkt wiederverwendet würde;
- Bei Medizinprodukten, die zur Verwendung mit anderen Produkten und/oder Allzweckgeräten bestimmt sind, müssen Informationen zur Identifizierung solcher Produkte oder Geräte enthalten sein, damit sie sicher zusammen verwendet werden können, und/oder
- Informationen zu bekannten Beschränkungen bei der Kombination von Produkten und Geräten;
- Bei Geräten, die Strahlung für medizinische Zwecke aussenden, müssen Angaben über Beschaffenheit, Art, Intensität und Verteilung der ausgesandten Strahlung sowie Informationen darüber, wie Anwender, Patienten oder Dritte zu schützen sind, enthalten sein;
- Bei implantierbaren Produkten die gesamten qualitativen und quantitativen Informationen über die Materialien und Substanzen, mit denen Patienten in Berührung kommen können;
- Bei implantierten Produkten Angaben zur Identifizierung des Produkts (Name, Seriennummer usw.), Warnungen und Vorsichtsmaßnahmen in Bezug auf gegenseitige Beeinflussung, medizinische Untersuchungen oder Umgebungsbedingungen, Informationen zur erwarteten Lebensdauer und sonstige Informationen zur Gewährleistung der sicheren Verwendung des Produkts.
- Detaillierte Anweisungen zur sicheren Entsorgung des Medizinprodukts, des Zubehörs und der Verbrauchsmaterialien.
- Eine Beschreibung, wann der Anwender einen Angehörigen der Gesundheitsberufe um Rat fragen sollte (falls das Produkt für Laien bestimmt ist)
- Für Produktgruppen ohne medizinischen Verwendungszweck, die in Anhang XVI der Verordnung aufgeführt sind (z. B. Kontaktlinsen und Fettabsaugungsgeräte), Angaben zum Fehlen eines klinischen Nutzens und zu den mit der Verwendung des Produkts verbundenen Risiken.
- Ein Hinweis, dass Anwender und/oder Patienten alle schwerwiegenden Vorkommnisse, die im Zusammenhang mit dem Produkt aufgetreten sind, dem Hersteller und der zuständigen Behörde des Landes melden müssen, in dem der Anwender und/oder Patient ansässig ist;
- Bei Produkten, zu deren Bestandteilen programmierbare Elektroniksysteme, einschließlich Software, gehören, oder Produkte in Form einer Software enthalten, müssen die Mindestanforderungen bezüglich Hardware, Eigenschaften von IT-Netzen und IT-Sicherheitsmaßnahmen, die für den bestimmungsgemäßen Einsatz der Software erforderlich sind, angegeben werden.
LADEN SIE DIE CHECKLISTE MIT ANFORDERUNGEN AN GEBRAUCHSANWEISUNGEN FÜR MEDIZINPRODUKTE HERUNTER
Sie finden die vollständige Übersicht in Kapitel III der Verordnung. Wenn Sie jedoch die gesamte Verordnung nach „Gebrauchsanweisung für Medizinprodukte“ durchsuchen, stellen Sie fest, dass diese Liste nicht vollständig ist.
An anderen Stellen finden Sie auch noch folgende Anforderungen:
- Die CE-Kennzeichnung muss in der Gebrauchsanweisung für Medizinprodukte (und auf jeder Verkaufsverpackung) angegeben sein.
- Für implantierbare Produkte und für Produkte der Klasse III muss der Hersteller einen Kurzbericht über Sicherheit und klinische Leistung erstellen. Der Hersteller muss in der Gebrauchsanweisung (oder auf dem Etikett) angeben, wo dieser Kurzbericht erhältlich ist. Der Inhalt des Kurzberichts ist in der Verordnung festgelegt.
Der Vollständigkeit halber schlage ich vor, dass Sie auch die Verordnung nach „Gebrauchsanweisung für Medizinprodukte“ durchsuchen.

Die Einhaltung der Medizinprodukteverordnung
Wo also passt die Gebrauchsanweisung in den Compliance-Prozess für Medizinprodukte? Lassen Sie uns dies in einen Kontext setzen.
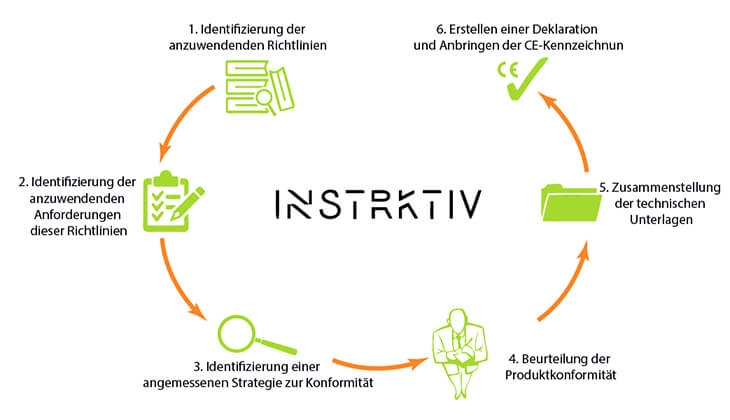
Um die Medizinprodukteverordnung einzuhalten, sollten Sie die sechs Schritte des CE-Kennzeichnungsverfahrens befolgen.
- Überprüfen Sie, ob Ihr Produkt unter die Verordnung fällt, und ermitteln Sie andere geltende Verordnungen, Richtlinien und Normen, die zur Sicherstellung der Compliance verwendet werden müssen.
- Stellen Sie fest, welche der gesetzlichen Anforderungen, die Sie in Schritt 1 gefunden haben, für Ihr spezifisches Produkt gelten.
- Stellen Sie fest, ob Ihr Medizinprodukt selbst bewertet werden kann oder ob Sie eine notifizierte Stelle benötigen. Für Produkte mit mittlerem bis hohem Risiko muss der Hersteller eine notifizierte Stelle beauftragen, um das Medizinprodukt zu bewerten und seine Konformität mit der Verordnung zu bestimmen. Bei Produkten mit geringem Risiko (Klasse I) ist die Bewertung eine Selbstzertifizierung.
- Bewerten Sie die Konformität des Produkts.
- Stellen Sie die technischen Unterlagen zusammen. Die technischen Unterlagen enthalten eine detaillierte Produktbeschreibung und -spezifikation gemäß Anhang II der Verordnung, das Etikett oder die Etiketten auf dem Produkt und seiner Verpackung, die Gebrauchsanweisung, Konstruktions- und Herstellungsinformationen, allgemeine Sicherheits- und Leistungsanforderungen, eine Nutzen-Risiko-Analyse sowie Informationen zum Risikomanagement und zur Produktverifizierung und -validierung.
- Bringen Sie die CE-Kennzeichnung an und erstellen Sie die Konformitätserklärung.
Die Anwendung harmonisierter Normen
Jedes Medizinprodukt, das in den Geltungsbereich der Medizinprodukteverordnung fällt, muss die Anforderungen dieser Verordnung erfüllen.
Diese Anforderungen sind sehr allgemeiner Natur, da sie für alle Medizinprodukte gelten.
Sie können harmonisierte Normen verwenden, um die Einhaltung der grundlegenden Anforderungen der Verordnung nachzuweisen. Für Produkte, die den Anforderungen harmonisierter Normen entsprechen, gilt die Konformitätsvermutung mit den entsprechenden wesentlichen Anforderungen.
Eine Typ-C-Norm enthält Spezifikationen für eine bestimmte Produktkategorie. Dies ist eine sogenannte vertikale Norm.
Es gibt harmonisierte Typ-C-Normen C für alle Arten von Medizinprodukten, wie z. B. Röntgengeräte, Operationsleuchten und Strahlentherapiesimulatoren, um nur einige zu nennen. Eine Übersicht der verfügbaren harmonisierten Normen finden Sie hier.
Diese Normen können zusätzliche oder detailliertere Anforderungen an die Gebrauchsanweisungen für Medizinprodukte enthalten.
Es wird empfohlen, zu überprüfen, ob für Ihren Produkttyp harmonisierte Normen existieren, und dementsprechend zu ermitteln, welche Anforderungen an Gebrauchsanweisungen darin enthalten sind.
Neben den vertikalen Normen gibt es auch horizontale Normen, die Sie anwenden können, um die Anforderungen der europäischen Gesetzgebung zu erfüllen.
Eine horizontale Norm ist eine Norm, die für alle Produktgruppen wie Maschinen, Spielzeug, Medizinprodukte und persönliche Schutzausrüstung gilt und nicht nur auf eine (vertikale) Produktkategorie beschränkt ist.
Die horizontale Norm für Gebrauchsanweisungen für Medizinprodukte ist die Norm IEC IEEE 82079-1:2019 (Hinweis: Diese Norm wurde zum Zeitpunkt der Veröffentlichung dieses Textes veröffentlicht, ist jedoch noch nicht Teil des Europäischen Rechtsrahmens, daher gilt bis dahin ihre Vorgängernorm, die ISO IEC 82079-1:2012).

Schauen wir uns das folgende Beispiel an, das den Unterschied zwischen einer allgemeinen Anforderung in einer Richtlinie/Verordnung und einer detaillierteren Anforderung in einer Norm zeigt.
Die Verordnung besagt, dass „die vom Hersteller bereitgestellten Informationen und Anweisungen für den Laien leicht verständlich und anwendbar“ sein müssen.
Die Norm 82079 enthält viele Anforderungen, anhand derer Sie sicherstellen können, dass Ihre Gebrauchsanweisung verständlich ist. Einige dieser Anforderungen sind:
- Anwendung des Minimalismusprinzips,
- Sicherstellung, dass die Gebrauchsanweisung für Medizinprodukte von der Zielgruppe hinsichtlich ihrer erwarteten Aufgaben und Ziele benutzbar und für diese Zielgruppe relevant ist;
- Verwendung einer einheitlichen Terminologie;
- Verwendung von für den Text geeigneten Schriftarten, Sicherheitszeichen und graphischen Symbolen, die für die Zielgruppe gut erkennbar und lesbar sind;
- Beauftragung kompetenter Personen mit der Erstellung der Gebrauchsanweisung.
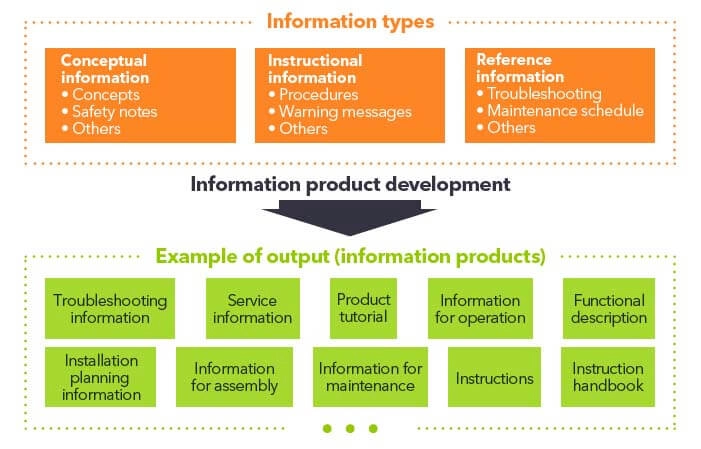
Welche Medien können Sie zur Veröffentlichung Ihrer Gebrauchsanweisung für Medizinprodukte verwenden?
In Europa ist für Produkte allgemein vereinbart, dass mindestens Sicherheitsanweisungen zusammen mit dem Produkt bereitgestellt werden sollten, d. h., dass alle nicht sicherheitsrelevanten Anweisungen online bereitgestellt werden können.
Wenn die Europäische Kommission die IEC 82079-1:2012 – Erstellen von Gebrauchsanweisungen – durch die IEC/IEEE 82079-1:2019 – Erstellen von Nutzungsinformationen – ersetzt, gelten höchstwahrscheinlich 2020 weniger strenge Anforderungen.
Warum?
Weil die neue Norm Folgendes festlegt:
„Der Lieferant bestimmt die Medien und das Format der Nutzungsinformationen gemäß der Art der Zielgruppen und auf der Grundlage ihrer Bedürfnisse.“
Verordnungen für Medizinprodukte waren jedoch schon immer ein Sonderfall.
In der Verordnung (EU) Nr. 207/2012 über elektronische Gebrauchsanweisungen für Medizinprodukte wurde bereits erwähnt, dass „das Vorliegen von Gebrauchsanweisungen für einige Medizinprodukte in elektronischer Form statt in Papierform ... für professionelle Nutzer von Vorteil sein“ könnte.
Diese Möglichkeit, die Gebrauchsanweisung nicht in Papierform, sondern in elektronischer Form zur Verfügung zu stellen, ist auf bestimmte Medizinprodukte beschränkt.
Eine Risikoanalyse sollte durchgeführt werden, um die Angemessenheit elektronischer Gebrauchsanweisungen festzustellen.
Wenn die folgenden Produkte ausschließlich für den professionellen Gebrauch bestimmt sind und die Verwendung durch andere Personen nicht vernünftigerweise vorhersehbar ist, können deren Gebrauchsanweisungen in elektronischer Form bereitgestellt werden:
- implantierbare Medizinprodukte und deren Zubehör, die ausschließlich zur Implantation oder Programmierung eines definierten aktiven implantierbaren Medizinprodukts bestimmt sind;
- fest installierte Medizinprodukte;
- Medizinprodukte und deren Zubehör, die mit einem eingebauten System ausgestattet sind, das die Gebrauchsanweisungen visuell anzeigt;
- eigenständige Software, die unter die Richtlinie 93/42/EWG fällt.

Sollten Sie Gebrauchsanweisungen für Medizinprodukte übersetzen?
Die schnelle Antwort lautet: ja!
Die ausführlichere Antwort:
Die Verordnung schreibt vor, dass die Gebrauchsanweisungen in die Sprache(n) übersetzt werden, die von den Mitgliedstaaten, in denen das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, akzeptiert werden.
Die meisten – wenn auch nicht alle – Mitgliedstaaten, verlangen, dass die Gebrauchsanweisungen übersetzt werden. Unter dem Gesichtspunkt der Produkthaftung ist es jedoch immer am besten, Gebrauchsanweisungen zu übersetzen.
Außerdem müssen das Etikett oder die Etiketten auf dem Produkt und seiner Verpackung in den Sprachen bereitgestellt werden, die von den Mitgliedstaaten, in denen das Medizinprodukt verkauft werden soll, akzeptiert werden.
Diese Übersetzungserfordernis gilt auch für die EU-Konformitätserklärung.
Was sind die Kennzeichnungspflichten für Medizinprodukte in Europa?
Obwohl es in diesem Beitrag um Gebrauchsanweisungen für Medizinprodukte geht, ist es meines Erachtens sinnvoll, auch die Anforderungen an die Kennzeichnung von Medizinprodukten kurz zu erörtern.
Die Kennzeichnung für Medizinprodukte müssen folgende Angaben enthalten:
- den Namen oder Handelsnamen des Produkts;
- alle unbedingt erforderlichen Angaben, aus denen der Anwender ersehen kann, worum es sich bei dem Produkt, dem Packungsinhalt sowie der Zweckbestimmung eines Produkts, sofern diese für den Anwender nicht offensichtlich ist, handelt;
- Name und Anschrift des Herstellers;
- wenn sich die Niederlassung des Herstellers außerhalb der EU befindet, Name und Anschrift des bevollmächtigten Vertreters;
- gegebenenfalls den Hinweis, dass das Produkt ein Arzneimittel (einschließlich eines Derivats aus menschlichem Blut oder Plasma), Gewebe oder Zellen menschlichen Ursprungs oder Gewebe oder Zellen tierischen Ursprungs enthält;
- gegebenenfalls nach Abschnitt 10.4.5 gekennzeichnete Angaben;
- die Losnummer oder die Seriennummer des Medizinprodukts;
- den UDI-Träger;
- eine eindeutige Angabe der Frist, innerhalb der das Produkt sicher implantiert werden kann;
- das Herstellungsdatum (falls keine Angabe des Datums, bis zu dem das Produkt sicher verwendet werden kann, vorhanden ist);
- gegebenenfalls einen Hinweis auf besondere Lagerungs- und/oder Handhabungsbedingungen;
- wenn das Medizinprodukt steril geliefert, einen Hinweis auf den sterilen Zustand und das Sterilisationsverfahren;
- Warnhinweise oder zu ergreifende Vorsichtsmaßnahmen;
- ist das Produkt für den einmaligen Gebrauch vorgesehen, einen Hinweis auf diesen Sachverhalt;
- falls es sich um ein aufbereitetes Produkt zum Einmalgebrauch handelt,
- einen Hinweis auf diesen Sachverhalt;
- die Anzahl der bereits durchlaufenen Aufbereitungszyklen;
- ögliche Beschränkungen hinsichtlich der Anzahl der Aufbereitungszyklen;
- bei einer Sonderanfertigung die Aufschrift „Sonderanfertigung“;
- einen Hinweis, dass es sich bei dem Produkt um ein Medizinprodukt handelt; Ist das Produkt lediglich für klinische Prüfungen vorgesehen, die Aufschrift „ausschließlich für klinische Prüfungen“;
- bei Produkten, die aus Stoffen oder aus Kombinationen von Stoffen bestehen, die dazu bestimmt sind, durch eine Körperöffnung oder durch Anwendung auf der Haut in den menschlichen Körper eingeführt zu werden, und die vom menschlichen Körper aufgenommen oder lokal im Körper verteilt werden, die qualitative Gesamtzusammensetzung des Produkts und quantitative Informationen zu dem(n) Hauptbestandteil(en), der (die) für das Erreichen der angestrebten Hauptwirkung verantwortlich ist (sind);
- bei aktiven implantierbaren Produkten die Seriennummer und bei anderen implantierbaren Produkten die Seriennummer oder die Losnummer;
Fazit
Medizinprodukte sind die am stärksten regulierten Produkte.
Auch die Gebrauchsanweisungen müssen sehr viele Anforderungen erfüllen.
Da im Internet viele unvollständige Informationen zu finden sind, habe ich diesen Beitrag darüber erstellt, wie man bessere Gebrauchsanweisungen für Medizinprodukte für zufriedenere und sicherere Anwender erstellt.
Erstellen Sie also nun Ihre Gebrauchsanweisung für Medizinprodukte oder schicken Sie uns eine E-Mail, wenn Sie dabei Unterstützung brauchen.
Sie können auch unsere Einweisung Medizinprodukte Vorlage erwerben.
Wie immer wäre es sehr schön, wenn Sie einen kurzen Kommentar hinterlassen würden, wenn dieser Beitrag für Sie nützlich gewesen ist!
 |
Ferry Vermeulen ist Experte für technische Kommunikation und Geschäftsführer von INSTRKTIV GmbH. Ferry hat es sich zur Aufgabe gemacht, digitale Benutzeranleitungen für alle Produkte der Welt zu erstellen. Hören Sie sich den INSTRKTIV-Podcast auf Spotify an oder lesen Sie einen seiner letzten Blogartikel. Xing I Spotify I YouTube I Facebook I Twitter |
MÖCHTEN SIE EINE VORLAGE FÜR EINE GEBRAUCHSANWEISUNG NUTZEN, DIE BEREITS ALLE RECHTLICHEN ASPEKTE ENTHÄLT?
Nehmen Sie den schnellsten Weg zu einer konformen Anleitung. Wir haben Vorlagen für Gebrauchsanweisungen für Medizinprodukte entwickelt, die bereits alle rechtlichen Inhalte beinhalten.