IFU for Medical Devices
25 May 2022 Law & Legislation
Creating instructions for use (IFU) for medical devices and in vitro diagnostic products requires quite a lot of responsibility.
In fact, you need to combine your techcomm skills with project management, administrative and legal skills. Luckily, what needs to be included in the instructions for use for medical devices has been determined in relevant legislation.
If there is any industry in which it is important to make sure that users can use devices safely, effectively and efficiently, it is the medical industry.
And let's not forget the importance of decreasing your liability by means of adequate instructions for use.
If you want to create a medical device user manual you need to be aware of both the content to include and the process to follow.
In this guide I’m going to show you everything you need to know when creating IFU for medical devices.
I will not only show you how to create IFU for the U.S. market, but also for the European market.
And I will show you for both markets what the requirements are for medical devices as well as for in vitro medical devices.
I hope the article provides sufficient medical device instructions for use guidelines and that it gives deep insight into relevant regulation on what you have to include in your IFU, in order to meet relevant EU regulations and/or the Code of Federal Regulations (CFR) in the U.S.
Understanding and applying the requirements will help you pass Notified Body conformity assessment (in Europe) and FDA approval in the U.S.
We’ll start with general labeling requirements in the U.S. If you want to go directly to IFU guidelines for the U.S. click here; for IFU requirements of in vitro devices in the U.S. click here.
For the IFU requirements on medical devices in Europe click here and for creating the IFU for in vitro devices for Europe click here.
Or use the below table of contents to navigate through the article.
But I recommend reading it all :)
Let’s dive right in!
DO YOU WANT A USER MANUAL TEMPLATE THAT ALREADY CONTAINS THE LEGAL PARTS?
Take the shortest way to a compliant manual. We have developed user manual templates for medical devices (EU and US) that contain all legal content.
Table of Contents
PART 1 - Instructions for Use (IFU) in the U.S.
What are the Requirements for IFU for Medical devices in the U.S.?
Apply the General Device Labeling Regulations for Medical Devices
Provide the name and address of the manufacturer on the label
Give a clear description of the intended uses
Provide adequate directions for use
Do not give misleading statements
Give label statements a prominent position
Use YYYY-MM-DD to format dates
Place a Unique Device Identifier (UDI) on the label
Apply Quality System Regulations to Create Adequate Instructions for Use (IFU)
Provide device identification information in the IFU
Provide handling information in the IFU
Provide storage information in the IFU
Provide installation information in the IFU
Make it easy to provide feedback
Provide servicing information in the IFU
Use the Guidance on Medical Device Patient Labeling
Include risk/benefit information and/or instructions for use.
Stimulate ease of navigation: include a Table of Contents
Include a glossary
Provide descriptive information
Provide operating instructions
Give clear setup instructions
Provide checkout instructions
Give operating instructions
Give accurate cleaning instructions
Give maintenance instructions
Give storage instructions
Give failure time information
Give disposal instructions
Provide information on accessories and other devices
Give troubleshooting information
Provide any additional information
Instruction for Use (IFU) Checklist
Apply FDA Approved Medical Device Standards
Apply labeling requirements for Electronic Products According to 21 CFR Part 100
Mark the device with a tag declaring compliance with applicable standards
Provide the name and address of the manufacturer
Provide the place, month and year of manufacture
Apply Labeling Requirements for Investigational Devices
Apply U.S. Labeling Requirements for In Vitro Diagnostic Products
PART 2 - Instructions for Use (IFU) in Europe
About the (EU) 2017/745 Regulation on Medical Devices
About the (EU) 2017/746 Regulation on In Vitro Medical Devices
Classification: List of Medical Devices Classes
List of Class I Medical Devices
List of Class IIa Medical Devices
List of Class IIb Medical Devices
List of Class III Medical Devices
What Information Should be Included in the Instructions for Use for Medical Devices?
What Information Should be Included in the Instructions for Use for In Vitro Medical Devices?
How to Comply with the (in vitro) Medical Device Regulation
How to Apply Medical Device IFU Standards
Who Can Draw Up the IFU for Medical Devices?
How to Use an Information Management Process to Develop Better Instructions for your Medical Device
Which Media Can You Use for Publishing Your IFU Medical Devices?
Should You Translate Instructions for Medical Devices?
IFU Medical Device Example
What are the Labelling Requirements for (in vitro) Medical Devices in Europe?
Conclusion
PART 1 - Instructions for Use (IFU) in the U.S.
In the U.S., laws are drafted by Congress.
Once Congress has enacted a law, the appropriate federal agency may create the regulations or rules to implement the law.
The different U.S. Federal Agencies regulating products and situations are divided into Hazards/Safety/Firearms, Vehicles/Vehicle-Related Products, Health/Body, and Other.
The Health and Body group oversees agencies that cover alcohol, tobacco, food, cosmetics and medical devices.
Safety of medical devices and in vitro diagnostic products in the U.S. is regulated by the FDA.
The Federal Food, Drug and Cosmetic Act (FFDCA) is the law under which the FDA takes action against regulated products.
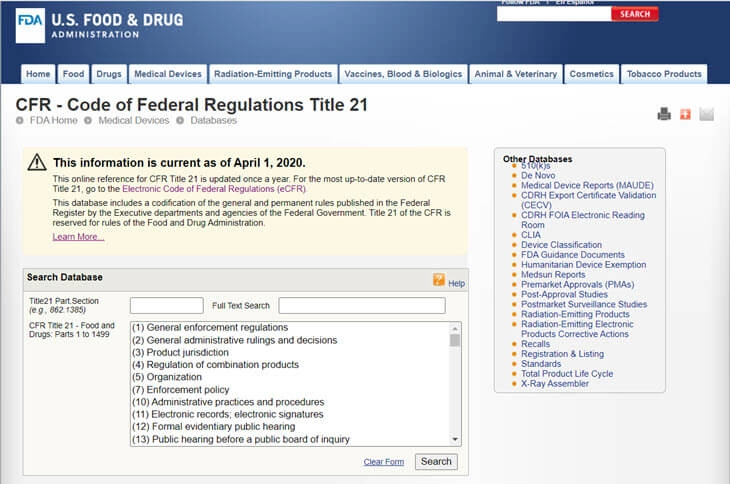
Based on the FFDCA, the FDA has created the CFR - Code of Federal Regulations Title 21. It contains the federal regulations for each product regulated by the FDA, including foods, pharmaceuticals, veterinary products and medical devices.
What are the Requirements for IFU for Medical Devices in the U.S.?
Instructions for Use (IFU) are part of the labeling regulations. CFR labeling requirements for medical devices give requirements on what to communicate on labels, and include things as the name and address of the manufacturer, intended use, directions for use, handling instructions, storage information, and installation information. The FDA’s guidance document contains more specific information on the instructions for use.and includes information on risk/benefit information, easy navigation, checkout instructions, monitoring informnation, cleaning instructions, and maintenance information.
I will discuss everything that falls under the CFR labeling requirements first. The FDA’s guidance document, will be discussed afterwards.
What falls under labeling?
Labeling’ is defined as 'all labels and other written, printed, or graphic matter (1) upon any article or any of its containers or wrappers, or (2) accompanying such article' at any time while a device is held for sale after shipment or delivery for shipment in interstate commerce.
Booklets, brochures, posters, tags, pamphlets, circulars, instruction books, direction sheets, fillers, etc. are also considered to fall under labeling. So the term 'accompanying' indicates that labeling is more than just the physical association with the product.

The labeling regulations for medical devices are found in several sections of Title 21 of the Code of Federal Regulations (CFR). This same overview can be found on the FDA’s website.
- General Device Labeling - 21 CFR Part 801
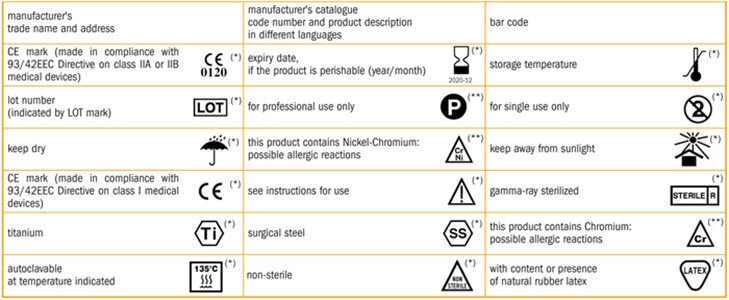
- Use of Symbols - 21 CFR Part 801.15
- In Vitro Diagnostic Products - 21 CFR Part 809
- General Electronic Products - 21 CFR Part 1010
- Unique Device Identification - 21CFR Part 830
- Good Manufacturing Practices - 21 CFR Part 820
- Investigational Device Exemptions - 21 CFR Part 812

In general, U.S. CFR labeling requirements for medical devices are more about what to communicate on labels, than containing requirements on what to put in the instructions for use.
That’s why I will discuss everything that falls under labeling in the next sections first.
The FDA’s guidance document, which I will discuss as well in this article, contains more specific information on the instructions for use.
Apply the General Device Labeling Regulations for Medical Devices
Part 801 of the CFR - Code of Federal Regulations Title 21 contains General Device Labeling requirements.
These requirements concern:
- Providing the name and address of the manufacturer
- The intended use
- The instructions for use
- Misleading statements
- Prominence of required label statements; use of symbols in labeling.
- Providing a Spanish-language version of certain required statements.
- How to format dates provided on a medical device label.
Let’s have a closer look at all of these requirements.
Provide the name and address of the manufacturer on the label
The label of a medical device in package form must state the name and place of business of the manufacturer, packer, or distributor.
When a corporation places the medical device on the market, only the actual corporate name shall be used. This name may be followed or preceded by the name of the specific division of the corporation.
Abbreviations, such as "Inc." etc., may be used and it is allowed to omit "The".
In cases where a medical device is not made by the entity whose name is put on the label, a phrase must be added that indicates the role of the entity, such as "Manufactured for ___" or "Distributed by _____".
The full business address must be given on the label: it must include the street address, city, State, and Zip Code.

Give a clear description of the intended uses
According to the CFR, the words intended uses refer to ‘the objective intent of the persons legally responsible for the labeling of devices.’
The IEC/IEEE 82079-1 standard describes the intended use as ‘an exhaustive range of functions or foreseen applications defined and designed by the supplier of the product.’
For example, the intended use of the Philips Ingenia MRI Scan is described as follows:

And the intended use of the Medtronic Model 5392 Pacemaker:

The intended use is what the device is developed for and says what it should be used for or how it can be used.
The intended use is often given in the instructions for use.
But also any claims on the device or on any product labeling, in advertisements or in oral or written statements are considered to be the intended use.
So always be careful what you communicate!
It is allowed to change the intended use of a device or product.
If, for example, a distributor sells a product that is intended to be used for a different purpose, then this distributor is allowed to do so.
In this case, the distributor, or seller, must provide adequate labeling in accordance with the new intended uses.
If a manufacturer is aware of any reasonably foreseeable misuse or use other than the new intended use, he/she must provide adequate labeling for such a device which clearly indicates how the product article should be used.
For example, the following screenshot, taken from a video, clearly states that the face mask sold by this distributor is not intended for medical purposes.

Provide adequate directions for use
It is considered to be important that a layperson receives adequate directions that enables him/her to use the medical device safely and according to its intended use.

There may be several reasons why directions are seen as inadequate.
Such as when the following information is (partly) missing or incorrect:
- Statements of all conditions, purposes, or uses for which such device is intended
- The quantity of the dose
- The frequency of administration or application
- The duration of administration or application
- The time of administration or application, in relation to time of meals, time when symptoms start, or other time factors.
- An adequate route or method of administration or application.
- Instructions on preparation for use, such as temperature adjustment, or other manipulation or process.
This example was found on the website of the FDA and represents an incorrect specification of the intended use

In this example a description of the intended use fails on the cover page:

Do not give misleading statements
A false or misleading representation with respect to another device, drug, food or cosmetic is considered to be a misleading statement.
Therefore, always make sure that the labeling of your product is authentic and does not look like other products.

Give label statements a prominent position
Section 502(c) of the FFDCA states that all required information should have a prominent position.

When there is a lack of prominence of a word, phrase or any other detail which must be displayed on the label, it may fall short of the FFDCA Act.
For example, the absence of the word, phrase, or detail in question on at least two sections of the label is considered to be a labeling defect.
The required statements must cover the entire area that is intended for placing the information.
Make sure that the label occupies adequate space on the product, and be visible enough to be clearly seen. Also, any other information shall not be more visible than the required information.
Apply a font size and type that is clearly legible and use sufficient contrast between the background and font color. Use a common, neutral design, that is not overwhelmed with other written or graphic content.
While there might be exemptions due to insufficiency of label space, no exemption will be considered if the label space was used for a phrase, word, detail device, or design that is deemed unnecessary or irrelevant under the FFDCA Act.
Also, in case of insufficiency of space, no other language shall be put on the label other than the English Language.
In regions where English is not the main language of communication, such as Puerto Rico, English may be replaced by the commonly spoken language of that region and when prescribed by a medical professional.
When symbols are used, they must be followed by a very descriptive and simplified note (for better understanding), written in the official language of the region.
A symbol can come without explanatory text, when it comes from an FDA approved standard, is used according to the FDA’s specifications for use and is explained in a symbol glossary.
A symbol developed by a Standards Development Organization, such as ISO, can also be used without an explanation, when:
- It is not in an FDA standard but not used according to the FDA’s specifications for use of the symbol;
- The manufacturer has determined that the symbol is likely to be understood;
- Is used according to the specifications for use as determined by the standardisation’s institute.
- Is explained in a symbol glossary.
Use YYYY-MM-DD to format dates
Dates included on a device’s label like expiry dates and production dates must have the approved YYYY-MM-DD formatting, with Y representing the year, M representing the month, and D representing the date.
Therefore, a product with expiry date March 26, 2022, for example, must look like this; 2014-03-26.
This is a typical guideline. These are cases of exemption though:
- Goods which have appropriate NDC (National Drug Code) numbers.
- Electronic products to which standards apply that are subject to the subchapter on Radiological health are exempted from the rule of YYYY-MM-DD date format mentioned above.

Place a Unique Device Identifier (UDI) on the label
Labels of all medical devices must bear a so-called Unique Device Identifier (UDI).
There are a few exceptions however.
Amongst others, finished devices manufactured and labeled prior to the compliance date established by the FDA, class I medical, that are exempted by the FDA from the good manufacturing practice requirements, some individual single-use devices, devices used solely for research, teaching, or chemical analysis(and not intended for any clinical use), veterinary medical device, custom devices, investigational device and devices intended for export do not need to bear the UDI.
The UDI must contain easily readable plain text and shall make use of automatic identification and data capture (AIDC) technology: a technology to automatically identify objects, collect data about them, and enter them directly into computer systems, without human involvement.
The UDI must include a lot or batch number, a serial number, a manufacturing date, an expiration date or any other element allowing the device’s identification.

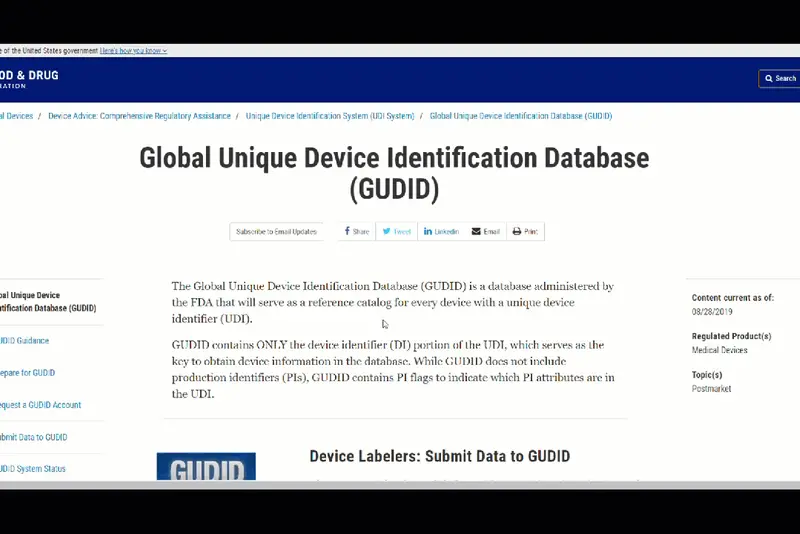
The FDA or FDA-accredited issuing agencies issue specific UDI systems. The UDI must be issued under one of these official systems.
The UDI must comply with ISO/IEC 15459-2, ISO/IEC 15459-4 and ISO/IEC 15459-6, and use only characters and numbers from the invariant character set of ISO/IEC 646.
Device data can be submitted to GUDID.
Stand-alone software may also be regulated as a medical device.
The UDI of stand-alone software that can be downloaded (so which is not packaged) must be displayed in easily readable plain-text whenever the software is started; or via a statement displayed through a menu command.

Apply Quality System Regulations to Create Adequate Instructions for Use (IFU)
The Quality System Regulations, sets forth ‘Current Good Manufacturing Requirements’ for the design, manufacture, packaging, labeling, storage, installation, and servicing of all finished medical devices intended for human use.
Compliance with the requirements, if applicable, must ensure that finished medical devices are safe and effective.
Indirectly, some requirements on the Instructions for use (IFU) can be found in this part of the CFR, although often well-hidden and not summed up in one specific IFU section.
Provide device identification information in the IFU
For example, section 820.60 contains requirements on device identification. It says:
Each manufacturer shall establish and maintain procedures for identifying products during all stages of receipt, production, distribution, and installation to prevent mixups.
This could mean that when a device is installed by following the installation instructions in the IFU, the installer should be able to identify the device.
This can be accomplished by putting a picture of the device on the cover of the product, including the product name, model, type, lot or batch number, or any other information to identify the product.

Provide handling information in the IFU
Section 820.140 gives requirements on providing device handling information.
It mentions that manufacturers must establish and maintain procedures to ensure that ‘mixups, damage, deterioration, contamination, or other adverse effects to the product do not occur during handling’.
Providing this information in the IFU is the most common thing to hand over this information.

Provide storage information in the IFU
In section 820.150 requirements on storage information are defined.
When storage information is needed to prevent adverse effects such as contamination, damage etc., this information shall be provided.
Typically, this information is also given to the user of the medical device through the instructions for use.


Provide installation information in the IFU
Section 820.170 gives requirements on the installation of devices.
When a device requires installation and/or a test procedure before it can be used, the manufacturer of the device must provide adequate installation, inspection and test procedure instructions.
Instructions and procedures must include instructions to ensure correct installation so that the device will perform as intended after installation.
These instructions and procedures must be provided with the device or made available to the person(s) installing the device in any other way.
It is the duty of the person installing the device, to make sure that the installation, inspection, and any required testing are conducted according to the provided instructions.
Basically, this means that you may want to provide some directional safety messages in your instructions for use.

Inspection and any test results shall be documented by the installer to demonstrate proper installation.
You may want to add a log book in your IFU to make this easier for the installer.

Make it easy to provide feedback
Section 820.180 gives requirements on keeping records. What does this mean?
It means, for example, that a manufacturer must keep a record that includes all device specifications, such as clear drawings, component specifications, software specifications, production process specifications and quality assurance procedures.
But also, packaging and labeling specifications and installation, maintenance, and servicing procedures and methods.
This is similar to the European requirement for establishing a technical file.
But also, complaints must be kept in the record. After all, the manufacturer has the duty to report certain events to the FDA.
A manufacturer must develop a procedure for receiving, reviewing and evaluating complaints. All complaints must be reviewed and evaluated to determine whether an investigation is necessary.
The instructions for use can facilitate an easy submission of complaints. There are several ways to do this.
You can include a section ‘Complaints’ (or any other meaningful name) which includes the company’s postal address, telephone number and email address.
When the information is available online as well, you may want to integrate a live chat, or submission form.
Things would be easier if information that should be collected in the record matches the information to be submitted.
This is the name of the device, the date the complaint was received, any unique device identifier (UDI) or universal product code (UPC), and any other device identification(s) and control number(s) used, the name, address, and phone number of the complainant and the nature and details of the complaint.

Provide servicing information in the IFU
Section 820.200 of the Quality System Regulations gives requirements on providing service information.
Often, servicing is required for the safe and efficient functioning of a medical device.
It is the responsibility of the manufacturer to provide instructions on how to service the device.
Service reports must be submitted to the manufacturer, who has accordingly the duty to analyze these.
When the service report represents an event which must be reported to the FDA, the manufacturer has to label the report as being a complaint and follow the procedure as described in the previous section.
Use the Guidance on Medical Device Patient Labeling
The document Guidance on Medical Device Patient Labelling, that has been published by the FDA, serves as a supporting document and describes in more detail what content to include in the instructions.
In my opinion, this guidance document is the best information source for creating IFU for medical devices for the U.S. market.
This guidance serves two purposes.
First of all, it should assist manufacturers in the development of the instructions for use.
Secondly, it assists FDA reviewers in their review and evaluation of medical device patient labeling.
The guidance aims to make medical device patient labeling more understandable to and usable by patients (or lay persons, such as family members caring for patients).
The guidance defines medical device patient labeling as “any information associated with a device targeted to the patient or lay caregiver”.

Medical device patient labeling is intended to help assure that a medical device is used effectively and safely.
The following formats of medical device labeling can be distinguished: user manuals, videos, brochures, leaflets, etc.
The guidance says the following about the content to include in the Medical Device Patient Labeling.
Include risk/benefit information and/or instructions for use.
Medical device labeling typically includes risk/benefit information and/or instructions for use (IFU).
Risk/benefit information can be included when patients or lay caregivers, for example, need to provide their care practitioner with personal health information.
Based on this information, the practitioner accordingly needs to decide whether to use or not use a certain device, or make a selection between available devices for the treatment, prevention, or diagnosis of an illness.
Also, risk/benefit information helps to determine what the possible effect of a medical device on the patient may be.

The development of instructions for use may be considered when patients or lay caregivers need to monitor and report on the operation or output, maintain or dispose of the device.
For example, diagnostic devices, such as a glucose monitor, mostly generate a certain output. The patient or caregiver needs to understand this output.
Also, when a device generates waste, has single use items, or contaminated reagents, these may be needed to be disposed of safely according to the instructions.
In many cases, both risk/benefit information and instructions for use have to be provided.
For example, when the user (patient or caregiver) needs to install, operate, interpret, and manipulate a medical device.
In those cases, it is often crucial to know how to use/operate the device safely. The user needs to read and understand any warnings, precautions, and contraindications, while working with the device.

Stimulate ease of navigation: include a Table of Contents
The guidance document stipulates the importance of a table of contents (ToC) for the sake of easy navigation.
The ToC is the spine of your user manual because it helps the user to quickly and easily find the information they need and helps them to find the answer to their questions.
A clear table of contents supports the user who is navigating to the right section, without having to frantically search through the entire instruction manual.
A table of contents should be included if the instructions for use are lengthy and/or complex.
The IEC/IEEE 82079 on Information for Use mentions that when instructions for use exceed 12 pages, they shall have a table of contents.

The standard recommends that the table of contents has enough levels to help the target audience find information efficiently, but emphasizes not to have more than four levels of headings.

Although not mentioned in the FDA Guidance document, page numbering and an index are also useful ways to help users navigate through the instructions for use (IFU).
When you have a dynamic content delivery (e.g. instructions in the form of online help), a search bar, links to related topics, individualized delivery and context sensitive delivery, these are ways that support navigation.
Include a glossary
Consistent terminology is crucial for clear understanding of the instructions for use and thus for the safe and effective use of the device..
Medical devices often come with their own specific jargon and terminology.
Make sure you explain the meaning of terminology used, including abbreviations, in a glossary.
This is especially important when the device is intended for use by a target audience without specialist knowledge.
If a glossary is used, place it directly after the table of contents. This will alert readers that it is there to help them.
Also, add any definitions to the text, or provide links and references.

Provide descriptive information
Besides instructional information, such as operation instructions and maintenance instructions, the IFU often needs to include descriptive information.
Descriptive information is information such as a description of the purpose of the device, a general description of the device, when the device should not be used (contraindications), risks and benefits information, expectations of the device and the procedure associated with the device and general warnings and precautions.
An illustration is often the best way to describe a device, parts and its accessories. The illustration can indicate all important parts that the user needs to be aware of in the rest of the IFU, such as displays, switches, dials, and meters.
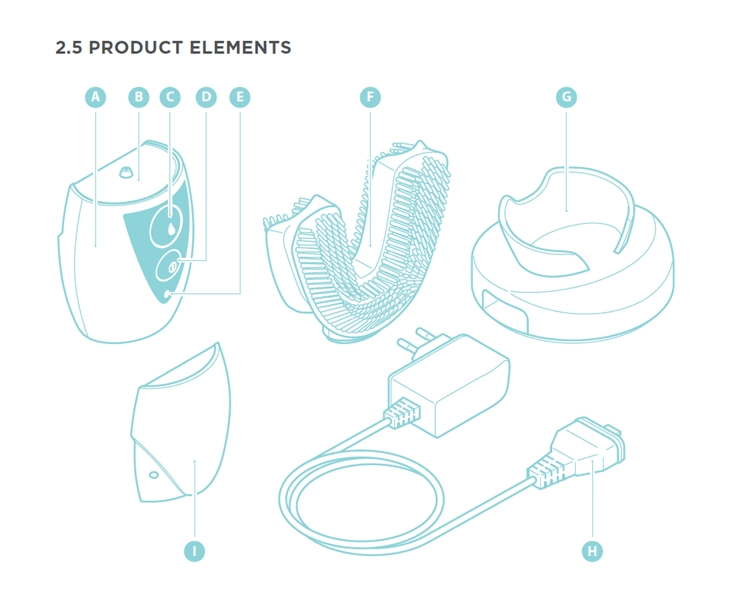
The function or purpose of each product element that contains a call out should be described in a table accompanying the illustration.
Also, include an overview of any materials in the device that may cause issues for patients with hypersensitivities.
Contraindications are conditions in which a medical device shall not be used. This is often because the risk outweighs any possible benefit.
For example, the medical device may be contraindicated for patients with epilepsy or for pregnant women.

As discussed previously, risk/benefit information can be included when patients or lay caregivers, for example, need to provide their care practitioner with personal health information.
Make sure that risk messages always provide a source for more information, acknowledge any uncertainties (e.g. indicate when there is a lack of currently available scientific knowledge), include analogies, discuss the nature of the risk, give alternatives and discuss benefits.
Benefit messages should provide balanced benefit information. It is not sensible to rely on using marketing waffle!
Therefore, benefit messages should identify outcomes, estimate the magnitude of outcomes in a way that is meaningful for readers to understand and evaluate the benefit for individuals.
General warnings and precautions are an integral part of risk and benefit information.

General warnings and precautions cover all specific hazard information that a user needs to be aware of before using a medical device.
You can best place the warnings and precautions at the beginning of the IFU. At least, you should act according to the guidance document….
The ANSI Z535.6 standard distinguishes directional safety instructions (such as “READ ALL INSTRUCTIONS BEFORE USE”), grouped safety messages, sectional safety messages and embedded warnings.

The first two are typically placed at the beginning. The other two respectively in the section or step to which they apply. The ANSI standard aims to warn the user of any risk that might occur.
I like that approach.
Appendix E of the Guidance document stresses this as well:
“Research shows that hazard alerts are most effective when integrated into the task information at the most relevant location.”
The appendix gives detailed additional information on definitions, purpose, content, format, location, etc related to warnings and precautions.

The appendix includes a remark about the importance of following the care regimen as explained in the IFU and how it helps to motivate the user to obey the instruction.
Regarding providing information on device expectations (and the procedure associated with the device), you may want to tell the patient what to expect before, during, and after a (surgical) procedure and/or the use of the medical device.
Consider giving instructions on post-procedural or post-operative care.
Provide operating instructions
Users need to know how to use a device. They should be given specific actionable instructions, placed in the right order.
Make clear which steps are crucial, and which ones are optional.
Besides marketing waffle, detailed technical descriptions of processes are also often unnecessary. Users care about solving their problems and finding answers to their questions: How do I run a test? How do I understand the score? How do I clean the mixer?
When writing the instructions, take into account that the user may not have medical knowledge or any experience with similar devices.
Using an eighth-grade writing style ensures that you reach most of the population.
Be clear, consistent and unambiguous!
Before giving the instruction to complete a task, start with stating the purpose and expected outcome of a task.

Give clear setup instructions
Let the user know if the setup of the device needs to be done by a qualified person. For example, by adding the following warning:
WARNING! Set up shall be carried out by a properly qualified and skilled installer.
Or
WARNING! Installation and commissioning shall be carried out by a properly qualified and skilled installer with due observance of the applicable rules and legislation.
If the lay user is allowed to set up the device, include the following information (if relevant):
- A parts list;
- A list of required tools and materials;
- Unpacking instructions;
- Instructions on how to recycle or dispose of packing waste;
- Information on the location of installation/placement, such as on a bench or floor.
- Set up related warnings or safety instructions. Make sure to embed them by placing them right before the corresponding task or instruction;
- Results of an incorrect setup;
- Numbered steps, containing setup instruction in a logical order;
- Information on any special preparations required before the first use of the device (such as cleaning or disinfecting information);
- Space for the layperson to add in user-specific instructions;
- Emergency telephone number.
Provide checkout instructions
Sometimes, a device requires checkout procedures for the sake of safety and effectiveness.
For example, this may be required directly after installation, to make sure that the device is properly installed.
Or after maintenance, to make sure that the device is ready for use again.
The below example is taken from a GE Healthcare manual and shows an example of checkout after installation.

Typically, a checkout procedure includes:
- When to conduct the checkout, such as after installation and/or after maintenance;
- Step-by-step instructions to check the proper functioning of the medical device;
- Troubleshooting information on what to do if the checkout shows that the device is not working properly;
- Technical support telephone number;
- A schedule indicating the correct frequency, times or days for when to check the device.
Give operating instructions
Operating instructions are some of the most crucial parts of the instructions for use. Make sure that these instructions are clear and easy-to-follow.
The operating instructions may include any information on special preparation the user needs before operation. Does he or she need to wash their hands, warm up the device or should the temperature remain between certain values?

Add any warnings or safety instructions specifically related to operation and again: place them immediately before the corresponding task or instruction.
If the user can recognize an incorrect operation, tell him/her how to recognize this.

As always, the operating steps should be numbered and placed in a logical sequence.
Tell the user what results to expect when an instruction has been executed. For example:

Or

Save some space in the IFU to add any user-specific instructions.
Provide a telephone number of the person to call if there is a problem.

Give instructions on how to monitor the device
For some devices it may be important to carefully monitor the correct functioning of the device.
If this is important to your specific medical device, emphasize this importance in the IFU.
Instruct the user how he/she can make sure that the device acts as intended.
Provide clear examples of what to check exactly and to make sure that the medical device still works properly.
For example, this is a very specific instruction given in order to monitor if a certain message appears on the GUI:

Give accurate cleaning instructions
As cleaning and sterilization of a medical device is often of huge importance for its safe use, cleaning instructions should be drafted with great care.
Always make sure to list all the supplies needed in a section specifically intended for step-by-step cleaning procedures.

Tell the user the recommended frequency of cleaning the device, what the cleaning accomplishes (e.g. reducing hazards) and what the results of failure to clean would be.
Include warnings and precautions for cleaning agents, explain what the consequences are of using inadequate cleaning solutions or methods and add instructions required for the correct disposal of the suggested cleaning agents.

Give maintenance instructions
Maintenance instructions give the user all necessary directions and information to maintain the device effectively.
They should clearly describe what maintenance actions are the responsibility of the user and what the frequency of those actions should be.
If certain maintenance instructions should be performed by qualified personnel only, this shall be indicated in the IFU.

Consider creating separate booklets if several users are involved for performing maintenance tasks, for example one booklet for the end-user of the device and one for the maintenance engineer.
Give storage instructions
Sometimes medical devices need to be stored for shorter or longer periods. The storage conditions may affect the device.
Provide storage instructions to prevent any malfunctioning or device damage.
Start with indicating to the user what the reason for proper storage is and what possible consequences are of improper storage.
Explain how the device needs to be properly prepared for storage and define the storage conditions.
When longer periods of storage affect the device, for example when there is a risk of leaking batteries, inform the user about these risks.
If appropriate, include information on extended storage in relevant sections, such as the installation/setup, checkout, operation, or maintenance section.
Give failure time information
Most devices have a limited lifetime. But even if they do not, it is always good to mention the technical lifespan in the IFU. Why?
Because this defines how long the device can be operated safely when the instructions in the IFU are being followed.
It limits the amount of information that has to be provided in the IFU.
For example, when the lifespan is 1 year, you won’t need to give any maintenance information on parts that need to be replaced after that year.
Defining the lifespan limits your responsibility, warranty and your liability.
You can also add information on the importance of following the instructions and what the results and expectations are when the device fails, or the instructions are not followed.
In case of failure, let the user know if it is dangerous (or safe) when the device fails and what the effect might be on the patient.
This information should also be part of the Risks/Benefits section.
Give disposal instructions
Not only waste, cleaning cloths, and single-use items, but also the device itself, should be disposed of in a proper way.
When appropriate, instruct the user how to safely dispose of the device.

Provide information on accessories and other devices
Medical devices often come with accessories, add-ons, additional items, extra features etc.
Use the IFU to list all appropriate content areas for each accessory. You can do this by including a separate section to describe any accessories, or by adding the information to the section to which it applies.
If relevant, put a general warning at the beginning of the instructions for use, advising users of problems that may occur if they use accessories other than those you recommend.
I often choose to add this information in the intended use section.

Give troubleshooting information
A proper troubleshooting section is crucial to help users solve any problems.
Typically, this is a separate section on the instructions for use.
The best way to present troubleshooting information, is by giving error information (what happens?), the cause (what is the problem?), and the solution (how do I solve the error?) in a 3-column table.
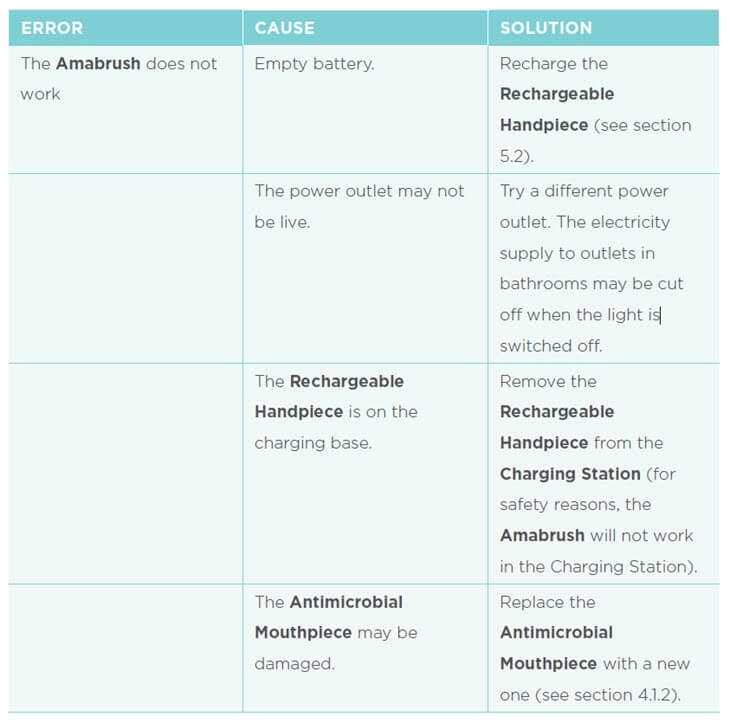
Note: The principles of Minimalism have another interesting way of providing troubleshooting information, called ‘supporting error recognition and recovery’, which is the 3rd Minimalism principle.
According to this principle, error recognition information and error solving information are provided within the instructions/steps, so not in a separate chapter, as in a troubleshooting chapter.
The idea behind this is that you want to help people to manage errors when they occur.
This means that you have to support detection and you have to support correction. These are the two minimalist aspects of error management.
The following example shows how error recognition and solving are embedded in the task domain.

Provide any additional information
Lastly, the guidance document for FDA reviewers recommends to include all kinds of additional information, such as clinical study information, disease and self-care information, adverse events, user assistance information, warranty information and travel or international use information.
It also mentions an index, which we discussed previously, and the date of printing.

Instruction for Use (IFU) Checklist
That is a lot of information that we have discussed. Therefore this checklist can be of use to make sure you include all information in the IFU:
- Table of Contents
- Glossary
- Purpose of the device (indications for use)
- Description of the device
- Contraindications
- Risks and benefits
- Expectations of the device and the procedure associated with the device
- Warnings and precautions
- Importance of the need to adhere to a care regimen
- Setup instructions
- Checkout procedure
- Operating instructions
- Monitoring instructions
- Cleaning instructions
- Maintenance instruction
- Storage instructions
- Expected failure time
- Disposal instructions
- Instructions on accessories and additional devices
- Troubleshooting Information:
- Clinical studies
- Disease and self-care information
- Adverse events
- arranty
- Travel or international use
- Index
- Date of Printing
- User Assistance Information

Apply FDA Approved Medical Device Standards
Most CFR requirements contain label requirements and regulate essential, but general, device information such as expiration date, intended use, dose quantity etc.
We have not found specific requirements on the IFU yet. Why?
Because it is hard to give all the requirements for each of the wide variety of devices that are available.
The contents of the IFU of a pacemaker are different from that of an MRI scan. The user of each device requires different information in order to use the device safely and effectively.
Standards are being developed to define requirements for specific medical devices or groups of medical devices.
For example, there are standards for wheelchairs and ambulatory electrocardiographic systems.
The same applies to dental cartridge syringes and steam sterilizers.
The FDA approves certain standards when they think the standards meet all safety and effectiveness goals of the FFDCA.
These can be national approved standards, such as the ANSI/ADA 48-2-2010 (R2015) LED Curing Lights.

Or international standards, such as ISO 10939:2017 for slit-lamp microscopes.

Approved standards can be found in the Federal Register.
Consult relevant approved standards, as they often include information on the IFU.
For example, the FDA approved standard IEC 61010-1 - Safety requirements for electrical equipment for measurement, control, and laboratory use - Part 1: General requirements contain requirements on the documentation.
Apply Labeling Requirements for Electronic Products According to 21 CFR Part 100
Now we’ve discussed the labeling requirements on all kinds of medical products, let’s have a closer look at electrical medical devices.
That’s because a medical device can also be an electronic device.
Actually, almost all medical products that we at INSTRKTIV deal with are electronic medical devices.
After all, electronics means that there are more features and complexity, so the more complex a medical device, the more instructions for use should come with the device.
See these two types of electronic medical devices:

Mark the device with a tag declaring compliance with applicable standards
A medical device must bear a tag or label containing certification and compliance information.
This tag or label shall be permanently fixed or inscribed.
It should have a prominent position, be legible, and easy to see when the product is ready for use.
Possibly, a mandatory standard may have its own requirements on the certification. In these cases, the standard shall be followed.
When one or more standards apply to an electronic device, the manufacturer must make sure that the dealer receives a certification that the device meets the applicable standard(s).
Provide the name and address of the manufacturer
The label of an electrical medical device must contain the name and address of the manufacturer. Abbreviations, such as “Co.” and "Inc." may be used.
When a medical device is sold under a different name other than the manufacturer, the full name and address of the entity under whose name the device is sold should be used, but only when this person or company has made the manufacturer’s name known to the Center for Devices and Radiological Health.
Also, this tag shall be permanently affixed to the device and be clearly visible and legible.
Provide the place, month and year of manufacture
As we’ve just seen that the date format of a medical article should have the form yy-mm-dd, for electronic devices, the place, month and year of manufacture should be mentioned.
The place of manufacture may take the form of a code, but only when this code has been made known to the Center for Devices and Radiological Health.
The month and year must be in a clear and legible format. The year must be shown as a four-digit number:
Manufactured: December 2020
Apply Labeling Requirements for Investigational Devices
When a medical device is either NOT approved for marketing in the U.S., or IS approved for marketing, but is being clinically evaluated for a new indication, a device is considered investigational.
Investigational devices are regulated and an Investigational Device Exemption (IDE) must be obtained from the FDA.
An IDE allows an investigational device to be used in order to collect safety and effectiveness data required to support a premarket approval (PMA) application or a premarket notification submission to the FDA.
All investigational devices must contain a label indicating the name and place of business of the manufacturer/packer/distributor, the quantity of contents (if appropriate), and the following statement:
CAUTION - Investigational device. Limited by Federal (or United States) law to investigational use.

The label or other labeling (such as the instruction for use) must give all relevant hazards, warnings, precautions, contraindications, adverse effects and interfering substances or devices.
Statements that are false or misleading in any particular way are strictly forbidden.
Among such statements are messages that the device is safe or effective for the purposes for which it is being investigated.
Investigational devices for laboratory animals must bear the following statement:
CAUTION - Device for investigational use in laboratory animals or other tests that do not involve human subjects.
Apply U.S. Labeling Requirements for In Vitro Diagnostic Products
In vitro diagnostic devices form a special group of medical devices and have their own set of requirements on the labeling and instructions for use (IFU).
In vitro diagnostic medical devices are tests used on biological samples to determine the status of a person's health.
According to the website of the FDA, in vitro diagnostic products are “those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.”
Section 809 of the Code of Federal Regulations (CFR) Title 21 applies to in vitro diagnostic products and Part 809.10 gives requirements on the labelling of in-vitro diagnostic products.
Requirements on the instructions for use (IFU) fall under the subsection ‘Labeling Requirements for Inserts and Outer Packaging’.
According to the CFR requirements, all required information must be in one place and in the format and order as specified in the requirements.
I have included this list below.
An exception is made when information is not applicable, or when a standard is available that requires differently.
Labels of reagents that are intended as a replacement in a diagnostic system, may be limited to contain only the information necessary to adequately identify the reagent and to describe its use in the system.
Multiple purpose instruments used for diagnostic purposes (and not committed to specific diagnostic procedures or systems) can be labeled with just the points annotated by an asterisk (*).
- * The proprietary and established product name;
- * The intended use of the product and whether it is a qualitative or quantitative type of procedure;
- Summary and explanation of the test, including a short history containing methodology and the special merits and limitations of the test;
- The chemical, physical, physiological, or biological principles of the procedure.

- For reagents:
- The common name, if any, and quantity, proportion, or concentration or each reactive ingredient; and for biological material, the source and measure of its activity;
- Appropriate cautions or warnings listed in 16 CFR Part 1500; the statement: "For In Vitro Diagnostic Use;" and any other limiting statements appropriate to the intended use of the product;
- Adequate directions for reconstitution, mixing, dilution, etc.;
- Appropriate storage instructions;
- A statement of purification or treatment required for use; and
- Physical, biological, or chemical indications of instability or deterioration.

- * For Instruments:
- Use or function;
- Installation procedures and requirements;
- Principles of operation;
- Performance characteristics and specifications;
- Operating instructions;
- Calibration procedures, including equipment and/or materials;
- Operational precautions and limitations;
- Hazards; and
- Service and maintenance information

- Specimen collection and preparation for analysis, describing;
- Special precautions/preparations;
- Additives necessary to maintain specimen integrity;
- Known interfering substances; and
- Recommended specimen storage, handling, and shipping instructions.
- A step by step outline of recommended procedures from the reception of the specimen to the obtaining of results. In addition to the following, this should include a list of any points that might improve precision or accuracy:
- A list of materials provided and instruction for use, e.g., reagents, equipment, etc.;
- A list of necessary materials that are not provided (include details such as sizes, numbers, types, and quality);
- A description of the amounts of reagents necessary, and parameters such as time, temperature etc.;
- A statement related to final reaction stability and any time restrictions on accurate measurements;
- Details of calibration, identifying and listing and necessary preparation of the reference materials, samples, and blanks. Describe the calibration range including the highest and lowest values measured; and
- Details of necessary quality control procedures and materials, e.g., positive and negative controls, acceptable performance limits.

- Explanation of the procedure for calculating the unknown, including the definition of each component of the formula, a sample calculation, and the number of significant figures appropriate for the answer;
- Limitations of the procedure, e.g., identify situations which will have an adverse impact on test results. If further testing, either more specific or more sensitive, is indicated, in all cases where certain results are obtained, the need for the additional test shall be stated;
- Expected values, including how the range(s) was established and identify the populations on which it was established;
- Specific performance characteristics as appropriate including accuracy, specificity, precision, and sensitivity;
- * Bibliography;
- * Name and place of business of the manufacturer, packer, or distributor; and
- * Date of issuance of the last labeling revision by the firm.

PART 2 - Instructions for Use (IFU) in Europe
All of the above information was about the requirements that the U.S. has on the instructions for medical devices. How are things in Europe organised?
A big difference is that in the U.S. the IFU falls under labeling requirements.
In the EU, the requirements on instructions, product marking and packaging are more clearly separated from each other.
Let’s have a look at the (EU) 2017/745 Regulation on medical devices and (EU) 2017/745 Regulation on in vitro devices.
About the (EU) 2017/745 Regulation on Medical Devices
The (EU) 2017/745 Regulation replaces the Medical Devices Directive 93/42/EEC and the Active Implantable Medical Devices Directive 90/385/EEC.
The new Medical Device Regulation will enter into force from May 26th 2021.
First of all, why has the directive become a regulation and what's the difference between the two?
A regulation has binding legal force throughout the member states of the EEA. A directive lays down certain results that must be achieved, but each individual member state is free to decide how directives will be transposed into national laws.
For example, in the Netherlands, labels and instructions for use must be in Dutch. This requirement is contained in Article 6 (2) of the Medical Devices Decree (Dutch: Besluit medische hulpmiddelen) and article 6 of the In-vitro Diagnostic Medical Devices Decree.
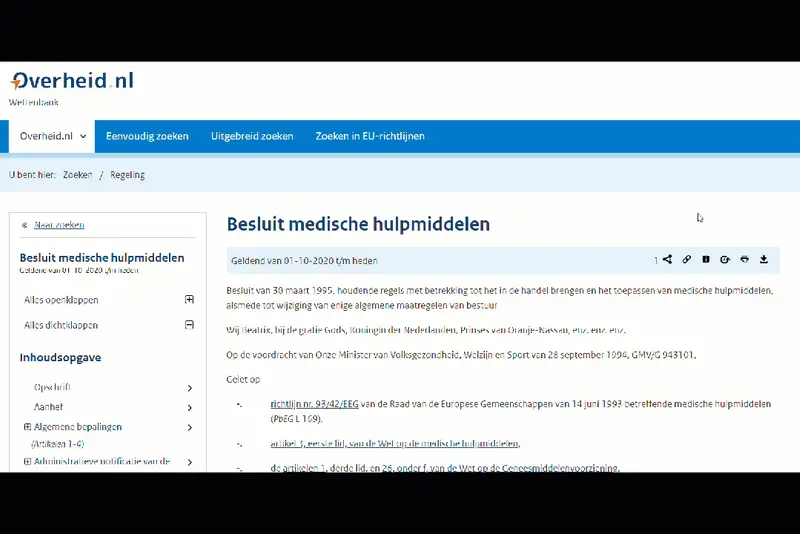
The example above describes how the Medical Device Directive is transposed into Dutch law. With the replacement of the directive by a regulation, those individual national laws will disappear.
The regulation harmonises the rules for the placing on the market and putting into service of medical devices and their accessories on the EU market.
The purpose of the regulation is to ensure the free movement of safe medical devices within the EU community.
But what exactly is a medical device in the EU?
A medical device is defined as any instrument, apparatus, appliance, software, implant, reagent, material or other item intended by the manufacturer to be used, alone or in combination, for human beings, for one or more of the following specific medical purposes:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations.

The regulation requires all these devices to:
- be suitable for their intended purpose;
- be safe and effective;
- not compromise the clinical condition or the safety of patients, or the safety and health of users.
The regulation also applies to accessories for medical devices and to certain product groups without a medical purpose, such as contact lenses, products intended to be totally or partially introduced into the human body through surgically invasive means, products intended for the purpose of modifying the anatomy or fixation of body parts, high intensity electromagnetic radiation emitting equipment and equipment intended for brain stimulation.
The regulation does not apply to in vitro diagnostic medical devices, medicinal products, cosmetic products, human blood, transplants and food.
In this podcast we talk more about How to CE mark medical devices:
About the (EU) 2017/746 Regulation on In Vitro Medical Devices
Also, Directive 98/79 on in vitro diagnostic medical devices will be replaced on May 26th 2021. The successor will be the Regulation (EU) 2017/746 on in vitro diagnostic medical devices.
The new regulation regulates in vitro diagnostic medical devices for human use and accessories for such devices in the EU.
So what exactly is called an in vitro diagnostic medical device in the EU?

An in vitro diagnostic medical device means ‘any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
- concerning a physiological or pathological process or state;
- concerning congenital physical or mental impairments;
- concerning the predisposition to a medical condition or a disease;
- to determine the safety and compatibility with potential recipients;
- to predict treatment response or reactions;
- to define or monitor therapeutic measures. Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices;
Classification: List of Medical Devices Classes
Medical devices have been divided into several classes by the (EU) 2017/745 Regulation:
- Class I
- Class IIb
- Class IIb
- Class III

A Notified Body must be involved in the assessment of classes IIa, IIb and III of medical devices as categorised by the Medical Device Regulation. The instructions for use are part of the conformity assessment procedure. They will be tested on completeness at least and sometimes also on correctness.
Instructions for use must be part of a medical device. By way of exception, instructions for use are not required for class I and class IIa devices if such devices can be used safely without any such instructions.
That's why the classification of your device also determines if and how you should provide your instructions.
Let's take a closer look at the classification.
List of Class I Medical Devices
Class I: These are the low risk medical devices. They include:
- Most non-invasive devices;
- Invasive devices intended for transient use;
- Reusable surgical instruments.
List of Class IIa Medical Devices
These are medium risk devices. They include:
- most invasive devices intended for short-term use;
- most surgically invasive devices intended for transient use;
- most active therapeutic devices intended to administer or exchange energy;
- most active devices intended for diagnosis and monitoring;
- most software intended to provide information which is used to make decisions about diagnosis or therapeutic purposes;
- most active devices intended to administer and/or remove medicinal products, body liquids or other substances to or from the body;
- most devices intended specifically to be used for disinfecting or sterilising medical devices;
- devices specifically intended for recording of diagnostic images generated by X-ray radiation;
- devices incorporating or consisting of nanomaterial if they present a negligible potential for internal exposure;
- most invasive devices with respect to body orifices, other than surgically invasive devices, which are intended to administer medicinal products by inhalation.
List of Class IIb Medical Devices
Class IIb devices are medium risk devices as well, but include:
- most invasive devices intended for long-term use;
- active devices intended to emit ionizing radiation for therapeutic purposes, including devices which control or monitor such devices, or which directly influence their performance;
- most devices used for contraception or prevention of the transmission of sexually transmitted diseases;
- all devices intended specifically to be used for disinfecting, cleaning, rinsing or, where appropriate, hydrating contact lenses, devices incorporating or consisting of nanomaterial if they present a low potential for internal exposure.

List of Class III Medical Devices
Class III devices are all high risk implantable devices. They include:
- devices intended specifically to control, diagnose, monitor or correct a defect of the heart or of the central circulatory system through direct contact with those parts of the body;
- active devices that are intended for controlling, monitoring or directly influencing the performance of active implantable devices;
- all devices incorporating, as an integral part, a substance which, if used separately, can be considered to be a medicinal product, as defined in point 2 of Article 1 of Directive 2001/83/EC, including a medicinal product derived from human blood or human plasma, as defined in point 10 of Article 1 of that Directive, and that has an action ancillary to that of the devices;
- most devices manufactured utilising tissues or cells of human or animal origin, or their derivatives, which are non- viable or rendered non-viable;
- devices incorporating or consisting of nanomaterial if they present a high or medium potential for internal exposure;
- active therapeutic devices with an integrated or incorporated diagnostic function which significantly determines the patient management by the device, such as closed loop systems or automated external defibrillators.

In vitro diagnostic devices have a different classification that groups devices into class A, B, C or D.
What Information Should be Included in the Instructions for Use for Medical Devices according to the Medical Device Regulation?
Most likely by now you know that you have a medical device that must include instructions for use. The next question would be: “what information do I need to put in my instructions?”
The Regulation includes the following (slightly reworded and restructured) list:
- The (trade) name of the medical device;
- The (trade) name (or registered trademark) and the address of the manufacturer;
- The date of issue of the instructions for use, or revision dates and information about where to find the latest revision of the instructions for use;
- If applicable, an indication that the medical device contains or incorporates a medicinal substance (including a human blood or plasma derivative), tissues or cells (or their derivatives of human origin), or tissues or cells of animal origin (or their derivatives)
- If applicable, information about substances that are subject to label requirements (according to section 10.4.5. of the Regulation);
- If the medical device is supplied sterile, an indication of its sterile state and the sterilisation method;
- If applicable, instructions for when a sterile packaging is damaged or accidently opened before use;
- Instructions for sterilisation (only applies to medical devices that are supplied non-sterile and should be sterilised before use);
- If applicable, a statement when the medical device is intended for single use;
- In the case of medical devices that are composed of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed by, or locally dispersed in the human body, the overall qualitative composition of the device, including quantitative information on the main constituent(s) responsible for achieving the principal intended action;
- The intended purpose, including any contra-indications, a specification of indications, the patient target group(s), and of the intended users;
- Training or qualification requirements and information about the device user and/or other persons and information about special facilities;
- Storing and handling information;
- Information that is needed to verify whether the medical device is properly installed and ready to be used as intended. This information must include, where relevant, instructions regarding maintenance, cleaning/disinfection, replacement of consumables, calibration and how to eliminate risks;
- Detailed information about all preparatory treatments or handling of the device before it is ready for use or during its use (e.g. sterilisation, calibration, final assembly). Also, the levels of disinfection required and the available methods for achieving these levels must be included.
- If applicable, the clinical benefits that can be expected;
- If applicable, links to the summary of safety and clinical performance (see article 32 of the Regulation);
- The performance characteristics of the medical device;
- If applicable, information allowing the healthcare professional to verify if the medical device is suitable to work with.
- If applicable, information allowing the healthcare professional to select the correct corresponding software and accessories;
- Any residual risks, contra-indications and any undesirable side-effects. This must include information to be conveyed to the patient.
- Any relevant warnings, precautions, contraindications, measures to be taken and limitations of use regarding the device.
- All relevant warnings and precautions when substances are intended to be introduced into the human body;
- Specifications the user requires to use the medical device appropriately, e.g. if the device has a measuring function, the degree of accuracy claimed for it must be given;
- For reusable devices, information on the processes for cleaning, disinfection, packaging and, if applicable, the validated method of re-sterilisation as determined by the country where the product is sold and used. When there are, for example, signs of degradation or in case the maximum number of allowable reuses has been reached, it must be mentioned that the device shall not be used any more;
- If applicable, a statement that a medical device can be reused when it is reconditioned under the responsibility of the manufacturer;
- Devices intended for single use only must contain information about the risks if the device were to be reused;
- Medical devices intended for use together with other devices and/or general purpose equipment must include information to identify such devices or equipment so they can be safely used together and/or information about any known restrictions to combinations of devices and equipment;
- For devices emitting radiation for medical purposes, detailed information about the nature, type and the intensity and distribution of the emitted radiation, and information about how to protect users, patients or other persons shall be given;
- For implantable devices, the overall qualitative and quantitative information about the materials and substances to which patients can be exposed;
- For implanted devices, information to identify the device (name, serial number etc), warnings and precautions regarding reciprocal interference, medical examinations or environmental conditions, information about the expected lifetime and any other information to ensure safe use of the device.
- Detailed instructions for the safe disposal of the medical device, its accessories and the consumables.
- A description of when the user should consult a healthcare professional (in case the device is intended for use by lay persons)
- For groups of products without an intended medical purpose that are listed in Annex XVI of the Regulation (such as contact lenses and liposuction equipment) information regarding the absence of a clinical benefit and the risks related to use of the device.
- A notice that users and/or patients must report any serious incident that has occurred in relation to the device to the manufacturer and the competent authority of the country in which the user and/or patient is established;
- For devices that incorporate electronic programmable systems (including software, or software that are devices in themselves) the minimum requirements concerning hardware, IT networks characteristics and IT security measures necessary to run the software as intended must be given.
DOWNLOAD THE IFU REQUIREMENTS FOR MEDICAL DEVICES

You can find this overview in full in Chapter III of the Regulation. However, when you scan the entire Regulation for 'instructions for use', you find that this list is not complete.
In other sections, the following requirements can be found as well:
- The CE marking must appear in the instructions for use (and on any sales packaging).
- For implantable devices and for class III devices, the manufacturer must draw up a summary of safety and clinical performance. The manufacturer must mention in the instructions (or on the label) where this summary is available. The content of the summary has been determined in the Regulation.
For the sake of completeness, I suggest that you ctrl + f the Regulation for 'instructions for use' as well.
Requirements on the instructions for use of in vitro diagnostic devices can be found here.
What Information Should be Included in the Instructions for Use for In Vitro Medical Devices according to the In Vitro Diagnostic Regulation (IVDR) Regulation?
The Regulation includes the following:
- the name or trade name of the device;
- necessary information to uniquely identify the device;
- the device's intended purpose:
- what is detected and/or measured;
- its function (e.g. screening, monitoring, diagnosis or aid to diagnosis, prognosis, prediction, companion diagnostic);
- the specific information that is intended to be provided in the context of:
- a physiological or pathological state;
- congenital physical or mental impairments;
- the predisposition to a medical condition or a disease;
- the determination of the safety and compatibility with potential recipients;
- the prediction of treatment response or reactions;
- the definition or monitoring of therapeutic measures;
- whether it is automated or not;
- whether it is qualitative, semi-quantitative or quantitative;
- the type of specimen(s) required;
- where applicable, the testing population; and
- for companion diagnostics, the International Nonproprietary Name (INN) of the associated medicinal product for which it is a companion test.
- an indication that the device is an in vitro diagnostic medical device, or, if the device is a ‘device for performance study’, an indication of that fact;
- the intended user, as appropriate (e.g. self-testing, near patient and laboratory professional use, healthcare professionals);
- the test principle;
- a description of the calibrators and controls and any limitation upon their use (e.g. suitable for a dedicated instrument only);
- a description of the reagents and any limitation upon their use (e.g. suitable for a dedicated instrument only) and the composition of the reagent product by nature and amount or concentration of the active ingredient(s) of the reagent(s) or kit as well as a statement, where appropriate, that the device contains other ingredients which might influence the measurement;
- a list of materials provided and a list of special materials required but not provided;
- for devices intended for use in combination with or installed with or connected to other devices and/or general purpose equipment:
- information to identify such devices or equipment, in order to obtain a validated and safe combination, including key performance characteristics, and/or
- information on any known restrictions to combinations of devices and equipment.
- an indication of any special storage (e.g. temperature, light, humidity, etc.) and/or handling conditions which apply;
- in-use stability which may include the storage conditions, and shelf life following the first opening of the primary container, together with the storage conditions and stability of working solutions, where this is relevant;
- if the device is supplied as sterile, an indication of its sterile state, the sterilisation method and instructions in the event of the sterile packaging being damaged before use;
- information that allows the user to be informed of any warnings, precautions, measures to be taken and limitations of use regarding the device. That information shall cover, where appropriate:
- warnings, precautions and/or measures to be taken in the event of malfunction of the device or its degradation as suggested by changes in its appearance that may affect performance,
- warnings, precautions and/or measures to be taken as regards the exposure to reasonably foreseeable external influences or environmental conditions, such as magnetic fields, external electrical and electromagnetic effects, electrostatic discharge, radiation associated with diagnostic or therapeutic procedures, pressure, humidity, or temperature,
- warnings, precautions and/or measures to be taken as regards the risks of interference posed by the reasonably foreseeable presence of the device during specific diagnostic investigations, evaluations, therapeutic treatment or other procedures such as electromagnetic interference emitted by the device affecting other equipment,
- precautions related to materials incorporated into the device that contain or consist of CMR substances, or endocrine disrupting substances or that could result in sensitisation or an allergic reaction by the patient or user,
- if the device is intended for single use, an indication of that fact. A manufacturer's indication of single use shall be consistent across the Union,
- if the device is reusable, information on the appropriate processes to allow reuse, including cleaning, disinfection, decontamination, packaging and, where appropriate, the validated method of re-sterilisation. Information shall be provided to identify when the device should no longer be reused, such as signs of material degradation or the maximum number of allowable reuses;
- any warnings and/or precautions related to potentially infectious material that is included in the device;
- where relevant, requirements for special facilities, such as a clean room environment, or special training, such as on radiation safety, or particular qualifications of the intended user;
- conditions for collection, handling, and preparation of the specimen;
- details of any preparatory treatment or handling of the device before it is ready for use, such as sterilisation, final assembly, calibration, etc., for the device to be used as intended by the manufacturer;
- the information needed to verify whether the device is properly installed and is ready to perform safely and as intended by the manufacturer, together with, where relevant:
- details of the nature, and frequency, of preventive and regular maintenance, including cleaning and disinfection;
- identification of any consumable components and how to replace them;
- information on any necessary calibration to ensure that the device operates properly and safely during its intended lifetime;
- methods for mitigating the risks encountered by persons involved in installing, calibrating or servicing devices.
- where applicable, recommendations for quality control procedures;
- the metrological traceability of values assigned to calibrators and control materials, including identification of applied reference materials and/or reference measurement procedures of higher order and information regarding maximum (self-allowed) batch to batch variation provided with relevant figures and units of measure;
- assay procedure including calculations and interpretation of results and where relevant if any confirmatory testing shall be considered; where applicable, the instructions for use shall be accompanied by information regarding batch to batch variation provided with relevant figures and units of measure;
- analytical performance characteristics, such as analytical sensitivity, analytical specificity, trueness (bias), precision (repeatability and reproducibility), accuracy (resulting from trueness and precision), limits of detection and measurement range, (information needed for the control of known relevant interferences, cross-reactions and limitations of the method), measuring range, linearity and information about the use of available reference measurement procedures and materials by the user;
- clinical performance characteristics as defined in Section 9.1 of this Annex;
- the mathematical approach upon which the calculation of the analytical result is made;
- where relevant, clinical performance characteristics, such as threshold value, diagnostic sensitivity and diagnostic specificity, positive and negative predictive value;
- where relevant, reference intervals in normal and affected populations;
- information on interfering substances or limitations (e.g. visual evidence of hyperlipidaemia or haemolysis, age of specimen) that may affect the performance of the device;
- warnings or precautions to be taken in order to facilitate the safe disposal of the device, its accessories, and the consumables used with it, if any. This information shall cover, where appropriate:
- infection or microbial hazards, such as consumables contaminated with potentially infectious substances of human origin;
- environmental hazards such as batteries or materials that emit potentially hazardous levels of radiation);
- physical hazards such as explosions.
- the name, registered trade name or registered trade mark of the manufacturer and the address of its registered place of business at which he/she can be contacted and its location be established, together with a telephone number and/or fax number and/or website address to obtain technical assistance;
- date of issue of the instructions for use or, if they have been revised, date of issue and identifier of the latest revision of the instructions for use, with a clear indication of the introduced modifications;
- (af)a notice to the user that any serious incident that has occurred in relation to the device shall be reported to the manufacturer and the competent authority of the member state in which the user and/or the patient is established;
- where device kits include individual reagents and articles that may be made available as separate devices, each of these devices shall comply with the instructions for use requirements contained in this section and with the requirements of this regulation;
- for devices that incorporate electronic programmable systems, including software, or software that are devices in themselves, minimum requirements concerning hardware, IT networks characteristics and IT security measures, including protection against unauthorised access, necessary to run the software as intended.
In addition, the instructions for use for devices intended for self-testing shall comply with all of the following principles:
- details of the test procedure shall be given, including any reagent preparation, specimen collection and/or preparation and information on how to run the test and interpret the results;
- specific particulars may be omitted provided that the other information supplied by the manufacturer is sufficient to enable the user to use the device and to understand the result(s) produced by the device;
- the device's intended purpose shall provide sufficient information to enable the user to understand the medical context and to allow the intended user to make a correct interpretation of the results;
- the results shall be expressed and presented in a way that is readily understood by the intended user;
- information shall be provided with advice to the user on action to be taken (in case of positive, negative or indeterminate result), on the test limitations and on the possibility of false positive or false negative result. Information shall also be provided as to any factors that can affect the test result such as age, gender, menstruation, infection, exercise, fasting, diet or medication;
- the information provided shall include a statement clearly directing that the user should not take any decision of medical relevance without first consulting the appropriate healthcare professional, information on disease effects and prevalence, and, where available, information specific to the member state(s) where the device is placed on the market on where a user can obtain further advice such as national helplines, websites;
- for devices intended for self-testing used for the monitoring of a previously diagnosed existing disease or condition, the information shall specify that the patient should only adapt the treatment if he has received the appropriate training to do so.
How to Comply with the (in vitro) Medical Device Regulation
So where do the instructions fit into the compliance process for Medical Devices? Let's put this into context.
In order to comply with the Medical Device Regulation, you should follow the six steps of the CE Marking process.
- Check whether your product falls under the Regulation and identify other applicable regulations, directives and standards to use in order to comply.
- Identify which requirements of the legislation that you found in step 1 apply to your specific product.
- Determine if your medical device can be self-assessed, or if you need a Notified Body. For medium to high risk devices, the manufacturer needs to hire a Notified Body to assess the medical device and determine its conformity to the regulation. For low risk devices (class I) the assessment is a self-certification.
- Assess the product’s conformity.
- Compile the technical file. The technical file includes a detailed device description and specification as determined in Annex II of the Regulation, the label or labels on the device and on its packaging, the instructions for use, design and manufacturing information, general safety and performance requirements, benefit-risk analysis and risk management and product verification and validation information.
- Affix the CE mark and draw up the declaration of conformity.
How to Apply Medical Device IFU Standards
Each medical device that falls within the scope of the Medical Device Regulation has to meet the requirements of the Regulation.
These requirements are very general in nature as they apply to all medical devices.
You can use harmonised standards to prove compliance with the essential requirements of the Regulation. Products that meet the requirements of harmonised standards benefit from a presumption of conformity with the corresponding essential requirements.
A C-type standard provides specifications for a given category of device. This is a so-called vertical standard.
There are C-type harmonised standards for all kinds of categories of medical devices, such as X-ray equipment, surgical luminaires and radiotherapy simulators, just to name a few. For an overview of available harmonised standards, see here and this PDF.
Some examples of standards are:
- EN 455-1:2000 Medical gloves for single use
- EN 1782:1998+A1:2009 Tracheal tubes and connectors
- EN ISO 10079-2:2009 Medical suction equipment
- EN ISO 11990:2018 Lasers and laser-related equipment
- EN 13060:2014 Small steam sterilizers
- EN 13976-2:2018 Rescue systems
- EN 14683:2019+AC:2019 Medical face masks - Requirements and test methods
- EN 60601-1-1:2001 Medical electrical equipment

Harmonised standards may contain additional, or more detailed, requirements for the instructions for use.

It is recommended to verify whether there are any harmonised standards for your device type and accordingly identify what requirements for the instructions these contain.
Besides the vertical standards, there are also horizontal standards that you can apply in order to comply with the requirements for the European legislation.
A horizontal standard is a standard that applies to all product groups, such as machinery, toys, medical devices and personal protective equipment, and is not limited to just one (vertical) product category.
The horizontal standard for Information for Use is the IEC/IEEE 82079-1:2019 standard (this standard is part of the European Legislation Framework).

Let's have a look at the following example that shows us the difference between a general requirement in a directive/regulation versus a more detailed requirement in a standard.
The regulation tells us that 'the information and instructions provided by the manufacturer shall be easy for the layperson to understand and apply'.
The 82079 standard gives many requirements for how you can assure that your instructions are understandable. Some of these requirements are:
- apply the principles of minimalism;
- make sure the information for use is usable and relevant for the target audiences with respect to their expected tasks and goals;
- use consistent terminology;
- use text fonts, safety signs and graphical symbols that are clearly legible for the target audiences;
- assign the creation of information for use to competent persons.
The 82079 also gives requirements for the font size. So if you want to have a guideline for the medical device ifu font size, use the sizes below.

Who Can Draw Up the IFU for Medical Devices?
I have given the answer already in the last bullet of the previous section: your instructions for use should be drawn up by a competent person.
So what is a competent person?
According to the 82079 standard, a competent person is 'a person who has acquired through training, qualifications or experience, or a combination of these, the knowledge and skills enabling that person to perform a specified task'.
The standard clearly describes the task-related competencies that the creation of information for use should include. These are, amongst others:
- conducting research and analysing the life cycle of a product;
- developing a content strategy;
- gathering safety-related information;
- developing content;
- validating the correctness of the content.
A company should identify the tasks to be performed to achieve the required results. They should evaluate the competencies which are needed to perform these tasks successfully and designate the tasks and responsibilities to persons who cover these competencies.
In some cases, only one person may be assigned to cover all responsibilities and tasks but in large organisations several persons, or maybe even a team or whole department, may be set up.
How to Use an Information Management Process to Develop Better Instructions for your Medical Device
When creating instructions for medical devices, you will find out that there is more to do than just technical writing.
You have to gather and analyse technical, marketing, compliance, legal and quality information. You have to structure and develop content and publish, distribute and maintain it,
Working closely with several departments is inevitable.
Each collaborator will have his or her specific ideas about how the instructions for use must be written to comply with regulations and meet marketing objectives.
An information management process can help you with this. Besides requirements for the instructions for use, the 82079 standard also gives requirements on the information management process.
The standard's starting point is that if you want to ensure that users can use a product safely, efficiently and effectively, your information management process should meet certain requirements.
The information management processes should be implemented for planning, designing, producing, and sustaining information for use.
The information management process requirements apply to all medical devices that are NOT consumer products.
The following four information management process groups are defined in the 82079 standard:
- analysis and planning of information;
- design and development (this includes reviewing, editing, and testing);
- production and distribution of information for use; and
- sustainment (including maintaining and improving the instructions).
Regarding the analysis and planning of information, the standard states that instructional procedures have to be formulated based on a market analysis, or an analysis of the characteristics, needs and intended tasks of the target audiences.
This analysis can include, amongst others, an analysis of the target audience (such as experience, background, language, skills, tasks, working environment and available tools), media, information sources, risk management, contractual agreements and legal considerations.
The development process shall include, for example, the preparation of information design concepts, templates, information gathering, selection of a content structure; editing and reviewing the content and usability testing.
The production and distribution process should include the integration, preparation, reproduction, packaging and distribution of physical media or electronic copies of the information for use.
And lastly, the sustainment, maintenance and improvement process includes continual target audience feedback and the establishment of a method for receiving information on changes, updating and making these updates available.
For more details about this, consult the 82079 standard as there is simply too much information to cover it all here.
Which Media Can You Use for Publishing Your IFU Medical Devices?
In Europe, it is generally agreed that safety instructions at least should be provided with the product, meaning that all non-safety related instructions can be provided online.
The EN IEC/IEEE 82079-1:2019 Preparation of Information for Use states the following on the medium:
‘The supplier shall determine the media and format of the information for use according to the nature of the target audiences and based on their needs’

However, regulations for medical devices have always been somewhat of an outlier.
Regulation (EU) No 207/2012 on electronic instructions for use of medical devices already mentioned that ‘for some medical devices the provision of instructions for use in electronic form instead of in paper form can be beneficial for professional users’. For most medical devices a print IFU must be provided.

This possibility of providing the instructions for use in electronic form instead of in paper form is limited to certain medical devices.
A risk analysis should be conducted to determine the appropriateness of electronic instructions for use.
When the following devices are intended for use exclusively by professional users and when the use by other persons is not reasonably foreseeable, their instructions may be provided in electronic form:
- implantable medical devices and their accessories intended to be used exclusively for the implantation or programming of a defined active implantable medical device;
- fixed installed medical devices ;
- medical devices and their accessories fitted with a built-in system visually displaying the instructions for use;
- stand-alone software covered by Directive 93/42/EEC.

Should You Translate Instructions for Medical Devices?
The short answer: yes!
The longer answer:
The Regulation requires that the instructions are translated into the language(s) accepted by the member states where the device is made available to the user or patient.
Most, if not all, member states require the instructions to be translated, but from a product liability point of view, it is always best to translate the instructions.
Also, the label or labels on the device and on its packaging must be provided in the languages accepted by the member states where the medical device is envisaged to be sold.
The translation requirement also applies to the EU declaration of conformity.
IFU Medical Device Example
If you are looking for a good example of an IFU for medical devices, try using our ifu medical device template.
Medical device instructions for use examples
What are the Labelling Requirements for (in vitro)
Medical Devices in Europe?
Although this post is about the IFU, I think it makes sense to briefly discuss the medical device labelling requirements as well.
After all, in the U.S., labelling and IFU are all pieces of the same cake.
Labels of medical devices in the EU shall bear the following:
- the name/trade name of the medical device;
- details necessary for a user to identify the device, the contents of the packaging and, where it is not obvious to the user, the intended purpose of the device;
- the name and address of the manufacturer;
- if the business is outside the European Union, the name and address of the authorised representative;
- where applicable, an indication that the device contains or incorporates a medicinal substance (including a human blood or plasma derivative), tissues or cells of human origin or tissues or cells of animal origin;
- where applicable, information labelled in accordance with section 10.4.5. of the regulation;
- the lot number or the serial number of the medical device;
- the UDI carrier
- an unambiguous indication of the time limit for using or implanting the device safely;
- the date of manufacture (only where there is no indication of the date until when it may be used safely);
- an indication of any special storage and/or handling conditions that apply;
- if the device is supplied sterile, an indication of its sterile state and the sterilisation method;
- warnings or precautions to be taken;
- if the device is intended for single use, an indication of that fact;.
- if the device is a single-use device that has been reprocessed:
- an indication of that fact;
- the number of reprocessing cycles already performed;
- any limitation as regards the number of reprocessing cycles.
- if the device is custom-made, the words ‘custom-made device’;
- an indication that the device is a medical device. If the device is intended for clinical investigation only, the words ‘exclusively for clinical investigation’;
- in the case of devices that are composed of substances or, of combinations of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed by or locally dispersed in the human body, the overall qualitative composition of the device and quantitative information on the main constituent or constituents responsible for achieving the principal intended action;
- for active implantable devices, the serial number, and for other implantable devices, the serial number or the lot number.
Labels of In Vitro Medical Devices label shall bear the following:
- the name or trade name of the device;
- the details strictly necessary for a user to identify the device and, where it is not obvious for the user, the intended purpose of the device;
- the name, registered trade name or registered trade mark of the manufacturer and the address of its registered place of business;
- if the manufacturer has its registered place of business outside the Union, the name of its authorised representative and the address of the registered place of business of the authorised representative;
- an indication that the device is an in vitro diagnostic medical device, or if the device is a ‘device for performance study’, an indication of that fact;
- the lot number or the serial number of the device preceded by the words LOT NUMBER or SERIAL NUMBER or an equivalent symbol, as appropriate;
- the UDI carrier as referred to in Article 24 and Part C of Annex VI;
- an unambiguous indication of the time limit for using the device safely, without degradation of performance, expressed at least in terms of year and month and, where relevant, the day, in that order;
- where there is no indication of the date until when it may be used safely, the date of manufacture. This date of manufacture may be included as part of the lot number or serial number, provided the date is clearly identifiable;
- where relevant, an indication of the net quantity of contents, expressed in terms of weight or volume, numerical count, or any combination of thereof, or other terms which accurately reflect the contents of the package;
- an indication of any special storage and/or handling condition that applies;
- where appropriate, an indication of the sterile state of the device and the sterilisation method, or a statement indicating any special microbial state or state of cleanliness;
- warnings or precautions to be taken that need to be brought to the immediate attention of the user of the device or to any other person. This information may be kept to a minimum in which case more detailed information shall appear in the instructions for use, taking into account the intended users;
- if the instructions for use are not provided in paper form in accordance with point (f) of Section 20.1, a reference to their accessibility (or availability), and where applicable the website address where they can be consulted;
- where applicable, any particular operating instructions;
- if the device is intended for single use, an indication of that fact. A manufacturer's indication of single use shall be consistent across the Union;
- if the device is intended for self-testing or near-patient testing, an indication of that fact;
- where rapid assays are not intended for self-testing or near-patient testing, the explicit exclusion hereof;
- where device kits include individual reagents and articles that are made available as separate devices, each of those devices shall comply with the labelling requirements contained in this section and with the requirements of this regulation;
- the devices and separate components shall be identified, where applicable in terms of batches, to allow all appropriate action to detect any potential risk posed by the devices and detachable components. As far as practicable and appropriate, the information shall be set out on the device itself and/or, where appropriate, on the sales packaging;
- the label for devices for self-testing shall bear the following particulars:
- the type of specimen(s) required to perform the test (e.g. blood, urine or saliva);
- the need for additional materials for the test to function properly;
- contact details for further advice and assistance.
Conclusion
Medical Devices and in vitro diagnostic devices are the most heavily regulated of products.
Also, the instructions have to fulfil quite a lot of requirements.
As there is a lot of incomplete information out there on the internet, I created this post for the sake of creating better information for use as well as happier and safer users.
So, go and create your IFU for medical devices yourself or send us an email if you are looking for support.
You can also purchase one of our instructions for use medical device template.
As always, leaving a short comment when this post is of use would be highly appreciated!
 |
Ferry Vermeulen is a technical communication expert and director at INSTRKTIV. It's Ferry’s mission to create digital user instructions for all products in the world. Listen to the INSTRKTIV podcast on Spotify or read one of his latest blog articles. Linkedin I Spotify I YouTube I Facebook I Twitter |
DO YOU WANT A USER MANUAL TEMPLATE THAT ALREADY CONTAINS THE LEGAL PARTS?
Take the shortest way to a compliant manual. We have developed user manual templates for medical devices (EU and US) that contain all legal content.